
Abstract
Multidrug-resistant tuberculosis (MDR-TB), caused by drug-resistant strains of Mycobacterium tuberculosis, is an increasingly serious problem worldwide. Here we examined a data set of whole-genome sequences from 5,310 M. tuberculosis isolates from five continents. Despite the great diversity of these isolates with respect to geographical point of isolation, genetic background and drug resistance, the patterns for the emergence of drug resistance were conserved globally. We have identified harbinger mutations that often precede multidrug resistance. In particular, the katG mutation encoding p.Ser315Thr, which confers resistance to isoniazid, overwhelmingly arose before mutations that conferred rifampicin resistance across all of the lineages, geographical regions and time periods. Therefore, molecular diagnostics that include markers for rifampicin resistance alone will be insufficient to identify pre-MDR strains. Incorporating knowledge of polymorphisms that occur before the emergence of multidrug resistance, particularly katG p.Ser315Thr, into molecular diagnostics should enable targeted treatment of patients with pre-MDR-TB to prevent further development of MDR-TB.
This is a preview of subscription content, access via your institution
Access options
Access Nature and 54 other Nature Portfolio journals
Get Nature+, our best-value online-access subscription
$29.99 / 30 days
cancel any time
Subscribe to this journal
Receive 12 print issues and online access
$209.00 per year
only $17.42 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout






Similar content being viewed by others

Accession codes
Primary accessions
BioProject
References
Farmer, P. et al. The dilemma of MDR-TB in the global era. Int. J. Tuberc. Lung Dis. 2, 869–876 (1998).
World Health Organization. Global Tuberculosis Report 2015 (World Health Organization, Geneva, Switzerland, 2015).
Awofeso, N. Antituberculosis medication side effects constitute major factor for poor adherence to tuberculosis treatment. Bull. World Health Organ. 86, B–D (2008).
Pietersen, E. et al. Long-term outcomes of patients with extensively drug-resistant tuberculosis in South Africa: a cohort study. Lancet 383, 1230–1239 (2014).
Boehme, C.C. et al. Rapid molecular detection of tuberculosis and rifampin resistance. N. Engl. J. Med. 363, 1005–1015 (2010).
Cohen, K. et al. Evolution of extensively drug-resistant tuberculosis over four decades revealed by whole-genome sequencing of Mycobacterium tuberculosis from KwaZulu-Natal, South Africa. PLoS Med. 12, e1001880 (2015).
Eldholm, V. et al. Four decades of transmission of a multidrug-resistant Mycobacterium tuberculosis outbreak strain. Nat. Commun. 6, 7119 (2015).
Izu, A., Cohen, T. & Degruttola, V. Bayesian estimation of mixture models with prespecified elements to compare drug resistance in treatment-naive and experienced tuberculosis cases. PLoS Comput. Biol. 9, e1002973 (2013).
Gegia, M., Winters, N., Benedetti, A., van Soolingen, D. & Menzies, D. Treatment of isoniazid-resistant tuberculosis with first-line drugs: a systematic review and meta-analysis. Lancet Infect. Dis. http://dx.doi.org/10.1016/S1473-3099(16)30407-8 (2016).
Biek, R. et al. Whole-genome sequencing reveals local transmission patterns of Mycobacterium bovis in sympatric cattle and badger populations. PLoS Pathog. 8, e1003008 (2012).
Blouin, Y. et al. Significance of the identification in the Horn of Africa of an exceptionally deep branching Mycobacterium tuberculosis clade. PLoS One 7, e52841 (2012).
Bryant, J.M. et al. Inferring patient-to-patient transmission of Mycobacterium tuberculosis from whole-genome sequencing data. BMC Infect. Dis. 13, 110 (2013).
Casali, N. et al. Evolution and transmission of drug-resistant tuberculosis in a Russian population. Nat. Genet. 46, 279–286 (2014).
Clark, T.G. et al. Elucidating emergence and transmission of multidrug-resistant tuberculosis in treatment-experienced patients by whole-genome sequencing. PLoS One 8, e83012 (2013).
Comas, I. et al. Out-of-Africa migration and neolithic coexpansion of Mycobacterium tuberculosis with modern humans. Nat. Genet. 45, 1176–1182 (2013).
Gardy, J.L. et al. Whole-genome sequencing and social-network analysis of a tuberculosis outbreak. N. Engl. J. Med. 364, 730–739 (2011).
Guerra-Assunção, J.A. et al. Recurrence due to relapse or re-infection with Mycobacterium tuberculosis: a whole-genome sequencing approach in a large, population-based cohort with a high HIV infection prevalence and active follow-up. J. Infect. Dis. 211, 1154–1163 (2015).
Merker, M. et al. Evolutionary history and global spread of the Mycobacterium tuberculosis Beijing lineage. Nat. Genet. 47, 242–249 (2015).
Perdigão, J. et al. Unraveling Mycobacterium tuberculosis genomic diversity and evolution in Lisbon, Portugal, a highly drug-resistant setting. BMC Genomics 15, 991 (2014).
Walker, T.M. et al. Whole-genome sequencing to delineate Mycobacterium tuberculosis outbreaks: a retrospective observational study. Lancet Infect. Dis. 13, 137–146 (2013).
Zhang, H. et al. Genome sequencing of 161 Mycobacterium tuberculosis isolates from China identifies genes and intergenic regions associated with drug resistance. Nat. Genet. 45, 1255–1260 (2013).
Winglee, K. et al. Whole-genome sequencing of Mycobacterium africanum strains from Mali provides insights into the mechanisms of geographic restriction. PLoS Negl. Trop. Dis. 10, e0004332 (2016).
Wollenberg, K.R. et al. Whole-genome sequencing of Mycobacterium tuberculosis provides insight into the evolution and genetic composition of drug-resistant tuberculosis in Belarus. J. Clin. Microbiol. http://dx.doi.org/10.1128/JCM.02116-16 (2016).
Gagneux, S. et al. Variable host–pathogen compatibility in Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. USA 103, 2869–2873 (2006).
Yimer, S.A. et al. Mycobacterium tuberculosis lineage 7 strains are associated with prolonged patient delay in seeking treatment for pulmonary tuberculosis in Amhara region, Ethiopia. J. Clin. Microbiol. 53, 1301–1309 (2015).
O'Reilly, L.M. & Daborn, C.J. The epidemiology of Mycobacterium bovis infections in animals and man: a review. Tuber. Lung Dis. 76 (Suppl. 1), 1–46 (1995).
Desjardins, C.A. et al. Novel d-cycloserine resistance mechanism in Mycobacterium tuberculosis revealed by whole-genome analysis. Nat. Genet. 48, 544–551 (2016).
Ma, Z., Lienhardt, C., McIlleron, H., Nunn, A.J. & Wang, X. Global tuberculosis drug development pipeline: the need and the reality. Lancet 375, 2100–2109 (2010).
Drummond, A.J., Suchard, M.A., Xie, D. & Rambaut, A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 29, 1969–1973 (2012).
Cattamanchi, A. et al. Clinical characteristics and treatment outcomes of patients with isoniazid-monoresistant tuberculosis. Clin. Infect. Dis. 48, 179–185 (2009).
Velayati, A.A. et al. High prevalance of rifampin-monoresistant tuberculosis: a retrospective analysis among Iranian pulmonary tuberculosis patients. Am. J. Trop. Med. Hyg. 90, 99–105 (2014).
Johnsson, K. & Schultz, P.G. Mechanistic studies of the oxidation of isoniazid by the catalase peroxidase from Mycobacterium tuberculosis. J. Am. Chem. Soc. 116, 7425–7426 (1994).
Rozwarski, D.A., Grant, G.A., Barton, D.H., Jacobs, W.R. Jr. & Sacchettini, J.C. Modification of the NADH of the isoniazid target (InhA) from Mycobacterium tuberculosis. Science 279, 98–102 (1998).
Quémard, A. et al. Enzymatic characterization of the target for isoniazid in Mycobacterium tuberculosis. Biochemistry 34, 8235–8241 (1995).
Marrakchi, H., Lanéelle, G. & Quémard, A. InhA, a target of the antituberculous drug isoniazid, is involved in a mycobacterial fatty acid elongation system, FAS-II. Microbiology 146, 289–296 (2000).
Sassetti, C.M., Boyd, D.H. & Rubin, E.J. Genes required for mycobacterial growth defined by high-density mutagenesis. Mol. Microbiol. 48, 77–84 (2003).
Bergval, I.L., Schuitema, A.R., Klatser, P.R. & Anthony, R.M. Resistant mutants of Mycobacterium tuberculosis selected in vitro do not reflect the in vivo mechanism of isoniazid resistance. J. Antimicrob. Chemother. 64, 515–523 (2009).
Ford, C.B. et al. Mycobacterium tuberculosis mutation rate estimates from different lineages predict substantial differences in the emergence of drug-resistant tuberculosis. Nat. Genet. 45, 784–790 (2013).
Bergval, I. et al. Pre-existing isoniazid resistance, but not the genotype of Mycobacterium tuberculosis, drives rifampicin resistance codon preference in vitro. PLoS One 7, e29108 (2012).
O'Sullivan, D.M., McHugh, T.D. & Gillespie, S.H. The effect of oxidative stress on the mutation rate of Mycobacterium tuberculosis with impaired catalase–peroxidase function. J. Antimicrob. Chemother. 62, 709–712 (2008).
Pym, A.S., Saint-Joanis, B. & Cole, S.T. Effect of katG mutations on the virulence of Mycobacterium tuberculosis and the implication for transmission in humans. Infect. Immun. 70, 4955–4960 (2002).
Salverda, M.L. et al. Initial mutations direct alternative pathways of protein evolution. PLoS Genet. 7, e1001321 (2011).
Ogbunugafor, C.B., Wylie, C.S., Diakite, I., Weinreich, D.M. & Hartl, D.L. Adaptive landscape by environment interactions dictate evolutionary dynamics in models of drug resistance. PLoS Comput. Biol. 12, e1004710 (2016).
Gumbo, T. et al. Concentration-dependent Mycobacterium tuberculosis killing and prevention of resistance by rifampin. Antimicrob. Agents Chemother. 51, 3781–3788 (2007).
Pasipanodya, J.G. & Gumbo, T. A new evolutionary and pharmacokinetic–pharmacodynamic scenario for rapid emergence of resistance to single and multiple antituberculosis drugs. Curr. Opin. Pharmacol. 11, 457–463 (2011).
Jutte, P.C., Rutgers, S.R., Van Altena, R., Uges, D.R. & Van Horn, J.R. Penetration of isoniazid, rifampicin and pyrazinamide in tuberculous pleural effusion and psoas abscess. Int. J. Tuberc. Lung Dis. 8, 1368–1372 (2004).
Diacon, A.H. et al. Early bactericidal activity of high-dose rifampin in patients with pulmonary tuberculosis evidenced by positive sputum smears. Antimicrob. Agents Chemother. 51, 2994–2996 (2007).
Long, M.W., Snider, D.E. Jr. & Farer, L.S. U.S. Public Health Service Cooperative trial of three rifampin–isoniazid regimens in treatment of pulmonary tuberculosis. Am. Rev. Respir. Dis. 119, 879–894 (1979).
Mitchison, D.A. Role of individual drugs in the chemotherapy of tuberculosis. Int. J. Tuberc. Lung Dis. 4, 796–806 (2000).
Mills, H.L., Cohen, T. & Colijn, C. Community-wide isoniazid preventive therapy drives drug-resistant tuberculosis: a model-based analysis. Sci. Transl. Med. 5, 180ra49 (2013).
Jindani, A. et al. High-dose rifapentine with moxifloxacin for pulmonary tuberculosis. N. Engl. J. Med. 371, 1599–1608 (2014).
Chien, J.Y. et al. Treatment outcome of patients with isoniazid-monoresistant tuberculosis. Clin. Microbiol. Infect. 21, 59–68 (2015).
Fasih, N., Rafiq, Y., Jabeen, K. & Hasan, R. High isoniazid resistance rates in rifampicin-susceptible Mycobacterium tuberculosis pulmonary isolates from Pakistan. PLoS One 7, e50551 (2012).
Jenkins, H.E., Zignol, M. & Cohen, T. Quantifying the burden and trends of isoniazid-resistant tuberculosis, 1994–2009. PLoS One 6, e22927 (2011).
Lumb, R. et al. Tuberculosis in Australia: bacteriologically confirmed cases and drug resistance, 2010. A report of the Australian Mycobacterium Reference Laboratory Network. Commun. Dis. Intell. Q. Rep. 37, E40–E46 (2013).
Nagu, T.J. et al. Multidrug- and other forms of drug-resistant tuberculosis are uncommon among treatment-naive tuberculosis patients in Tanzania. PLoS One 10, e0118601 (2015).
Villegas, L. et al. Prevalence, risk factors and treatment outcomes of isoniazid- and rifampicin-monoresistant pulmonary tuberculosis in Lima, Peru. PLoS One 11, e0152933 (2016).
Espinal, M.A. et al. Standard short-course chemotherapy for drug-resistant tuberculosis: treatment outcomes in six countries. J. Am. Med. Assoc. 283, 2537–2545 (2000).
Menzies, D. et al. Standardized treatment of active tuberculosis in patients with previous treatment and/or with monoresistance to isoniazid: a systematic review and meta-analysis. PLoS Med. 6, e1000150 (2009).
Huyen, M.N. et al. Epidemiology of isoniazid-resistance mutations and their effect on tuberculosis treatment outcomes. Antimicrob. Agents Chemother. 57, 3620–3627 (2013).
Denkinger, C.M., Pai, M. & Dowdy, D.W. Do we need to detect isoniazid resistance in addition to rifampicin resistance in diagnostic tests for tuberculosis? PLoS One 9, e84197 (2014).
Bai, Y., Wang, Y., Shao, C., Hao, Y. & Jin, Y. GenoType MTBDRplus assay for rapid detection of multidrug resistance in Mycobacterium tuberculosis: a meta-analysis. PLoS One 11, e0150321 (2016).
Department of Health, South Africa. Management of Drug-Resistant Tuberculosis: Policy Guidelines http://www.health-e.org.za/wp-content/uploads/2014/06/MDR-TB-Clinical-Guidelines-Updated-Jan-2013.pdf (2013).
Banerjee, A. et al. inhA, a gene encoding a target for isoniazid and ethionamide in Mycobacterium tuberculosis. Science 263, 227–230 (1994).
Larsen, M.H., Biermann, K., Tandberg, S., Hsu, T. & Jacobs, W.R. Jr. Genetic manipulation of Mycobacterium tuberculosis. Curr. Protoc. Microbiol. 6, 10A.2 (2007).
Walker, B.J. et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome-assembly improvement. PLoS One 9, e112963 (2014).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Salazar, A., Earl, A., Desjardins, C. & Abeel, T. Normalizing alternate representations of large sequence variants across multiple bacterial genomes. BMC Bioinformatics 16, 116 (2015).
Price, M.N., Dehal, P.S. & Arkin, A.P. FastTree: computing large minimum evolution trees with profiles instead of a distance matrix. Mol. Biol. Evol. 26, 1641–1650 (2009).
Walker, T.M. et al. Whole-genome sequencing for prediction of Mycobacterium tuberculosis drug susceptibility and resistance: a retrospective cohort study. Lancet Infect. Dis. 15, 1193–1202 (2015).
Farhat, M.R. et al. Genomic analysis identifies targets of convergent positive selection in drug-resistant Mycobacterium tuberculosis. Nat. Genet. 45, 1183–1189 (2013).
Swofford, D. PAUP: Phylogenetic Analysis Using Parsimony, Version 3.0 (Illinois Natural History Survey, 1989).
Guerra-Assunção, J.A. et al. Large-scale whole-genome sequencing of M. tuberculosis provides insights into transmission in a high-prevalence area. eLife 4, e05166 (2015).
Roetzer, A. et al. Whole-genome sequencing versus traditional genotyping for investigation of a Mycobacterium tuberculosis outbreak: a longitudinal molecular epidemiological study. PLoS Med. 10, e1001387 (2013).
Ford, C.B. et al. Use of whole-genome sequencing to estimate the mutation rate of Mycobacterium tuberculosis during latent infection. Nat. Genet. 43, 482–486 (2011).
Acknowledgements
We would like to thank the Broad Institute's Genome Sequencing Platform and Assembly and Annotation teams, including S.K. Young, M.E. Priest, T.P. Shea, B.J. Walker, L. Alvarado, M.G. Fitzgerald, S. Gujja, S. Hamilton, C. Howarth, J.D. Larimer, M.D. Pearson, Q. Zeng and J. Wortman. We would like to thank J. Romano and A. Keo for help with lineage detection, and M. Zambrano, B. Ferro and J.C. Rozo for isolation and phenotypic characterization of strains. We are also grateful to members of the TBResist Consortium for contribution of their strains, phenotypic data and expertise, and their help in forging collaborations, and to V. Dartois, D. Thomas, D. Hung and D. Plachetzki for helpful conversations. We also thank three anonymous reviewers of our manuscript for their insights and helpful suggestions. This project has been funded in whole or in part with federal funds from the National Institute of Allergy and Infectious Diseases (NIAID), National Institutes of Health, Department of Health and Human Services (grant U19AI110818 to the Broad Institute (B.W.B. and A.M.E.)), contract HHSN272200900018C to the Broad Institute (B.W.B.) and contract HHSN2722000900050C to the TB Clinical Diagnostics Research Consortium, the Intramural Research Program of NIAID (C.E.B. and L.E.V.) and the Korean CDC, Korean Ministry of Health and Welfare. This work was also funded (in part) by the intramural research program of the NIAID, NIH (C.E.B.). Funding was also provided by NIH grant 5U01AI069924-07 for IeDEA (A.S.P.), the Howard Hughes Medical Institute (W.R.B.) and NIH grant R01 AI110386 for 'Host–pathogen interactions in a failing global lineage of MTBC: M. africanum' (W.R.B.). The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of the NIH.
Author information
Authors and Affiliations
Consortia
Contributions
A.L.M., K.A.C., T.A., C.A.D., B.W.B. and A.M.E. conceived the project; A.L.M., K.A.C., T.A., C.A.D. and A. Salazar analyzed the data; A.L.M., K.A.C. and A.M.E. interpreted results; A.L.M. and K.A.C. wrote the manuscript; and D.T.A., C.E.B., J.B., S.B.C., S.-N.C., A.G., J.G., A.M.J., M.J., P.J., J.S.L., L.M., M.M., D.N., E.N., E.R., A. Skrahina, W.S., A.A.V., K.W., A.Z., L.E.V., G.H.C., S.E.D., J.E., P.F., J.E.G., A.R., V.C., D.H., P.-R.H., S.N., A.S.P., S.S., M.V.d.W., D.A., W.R.B., T.C. and S.H. were involved in sample acquisition and handling, including oversight of these activities. All authors critically read and revised the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Integrated supplementary information
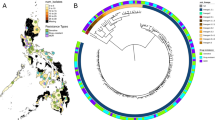
Supplementary Figure 1 Lineage and geographic distribution of M. tuberculosis isolates.
(a) Geographic distribution of M. tuberculosis isolates by lineage. This plot shows the sampling frequency within our data set of 5,310 strains and is not necessarily indicative of overall global incidence. The pie charts illustrate the relative proportions of M. tuberculosis lineages isolated within each of 11 UN subregions (http://unstats.un.org/unsd/methods/m49/m49regin.htm). The map is colored by UN geographic subregion. There were no strains in our data set from geographic regions colored gray. UN geographic subregions with fewer than 30 strains were excluded from this figure. This map was modified from a blank map of UN geographical subregions (Pusch, T.; licensed under CC BY-SA 3.0 via Wikimedia Commons; https://commons.wikimedia.org/wiki/File:Geografiaj_subregionoj_la%C5%AD_Unui%C4%9Dintaj_Nacioj_malplene.svg). (b) Global prevalence for each of six WHO global subregions as a percentage of total global TB burden (black bars), side by side with the percentage of our data set from each of these regions. We used different geographic subdivisions (WHO rather than UN) for this panel to present our data side by side with WHO data. Note that our data set is under-represented in strains from Southeast Asia (which includes India) and over-represented in strains from Africa and Europe (which includes Russia). WHO global subregions are defined as follows. AFR (Africa): Algeria, Angola, Benin, Burkina Faso, Cameroon, Cape Verde, Chad, Comoros, Equatorial Guinea, Gabon, Gambia, Ghana, Guinea, Guinea-Bissau, Liberia, Madagascar, Mali, Mauritania, Mauritius, Niger, Nigeria, Sao Tome and Principe, Senegal, Seychelles, Sierra Leone, Togo, Botswana, Burundi, Central African Republic, Congo, Côte d'Ivoire, Democratic Republic of the Congo, Eritrea, Ethiopia, Kenya, Lesotho, Malawi, Mozambique, Namibia, Rwanda, South Africa, Swaziland, Uganda, United Republic of Tanzania, Zambia, Zimbabwe; AMR (Americas): Canada, Cuba, United States of America, Antigua and Barbuda, Argentina, Bahamas, Barbados, Belize, Brazil, Chile, Colombia, Costa Rica, Dominica, Dominican Republic, El Salvador, Grenada, Guyana, Honduras, Jamaica, Mexico, Panama, Paraguay, Saint Kitts and Nevis, Saint Lucia, Saint Vincent and the Grenadines, Suriname, Trinidad and Tobago, Uruguay, Venezuela, Bolivia, Ecuador, Guatemala, Haiti, Nicaragua, Peru; EMR (Eastern Mediterranean): Bahrain, Cyprus, Iran (Islamic Republic of), Jordan, Kuwait, Lebanon, Libyan Arab Jamahiriya, Oman, Qatar, Saudi Arabia, Syrian Arab Republic, Tunisia, United Arab Emirates, Afghanistan, Djibouti, Egypt, Iraq, Morocco, Pakistan, Somalia, Sudan, Yemen; EUR (Europe): Andorra, Austria, Belgium, Croatia, Czech Republic, Denmark, Finland, France, Germany, Greece, Iceland, Ireland, Israel, Italy, Luxembourg, Malta, Monaco, Netherlands, Norway, Portugal, San Marino, Slovenia, Spain, Sweden, Switzerland, United Kingdom, Albania, Armenia, Azerbaijan, Bosnia and Herzegovina, Bulgaria, Georgia, Kyrgyzstan, Poland, Romania, Slovakia, Tajikistan, The Former Yugoslav Republic of Macedonia, Turkey, Turkmenistan, Uzbekistan, Yugoslavia, Belarus, Estonia, Hungary, Kazakhstan, Latvia, Lithuania, Republic of Moldova, Russian Federation, Ukraine; SEAR (Southeast Asia): Indonesia, Sri Lanka, Thailand, Bangladesh, Bhutan, Democratic People's Republic of Korea, India, Maldives, Myanmar, Nepal, Timor Leste; WPR (Western Pacific): Cambodia, China, Cook Islands, Fiji, Kiribati, Lao People's Democratic Republic, Malaysia, Marshall Islands, Micronesia (Federated States of), Mongolia, Nauru, Niue, Palau, Papua New Guinea, Philippines, Republic of Korea, Samoa, Solomon Islands, Tonga, Tuvalu, Vanuatu, Viet Nam, Australia, Brunei Darussalam, Japan, New Zealand, Singapore.
Supplementary Figure 2 Phylogenetic tree of all 5,310 M. tuberculosis isolates indicating data set of origin.
Phylogenetic tree of all 5,310 M. tuberculosis isolates (Online Methods). Colors in the outer circle indicate the data set of origin for each strain. In the central radial phylogeny, lineages are labeled and color-coded as follows: pink, lineage 1; blue, lineage 2; purple, lineage 3; red, lineage 4; brown, lineage 5 (M. africanum); dark green, lineage 6 (M. africanum); orange, lineage 7; light green, M. bovis.
Supplementary Figure 3 Phylogenetic tree of all 5,310 M. tuberculosis isolates indicating geographic region of isolation.
Phylogenetic tree of all 5,310 M. tuberculosis isolates (Online Methods). Colors in the outer circle indicate the geographic region of isolation. In the central radial phylogeny, lineages are labeled and color-coded as follows: pink, lineage 1; blue, lineage 2; purple, lineage 3; red, lineage 4; brown, lineage 5 (M. africanum); dark green, lineage 6 (M. africanum); orange, lineage 7; light green, M. bovis.
Supplementary Figure 4 Phylogenetic tree of all 5,310 M. tuberculosis isolates indicating genotypic drug resistance pattern.
Phylogenetic tree of all 5,310 M. tuberculosis isolates (Online Methods). Colors in the outer circle indicate the genotypic drug resistance pattern of the corresponding strain. In the central radial phylogeny, lineages are labeled and color-coded as follows: pink, lineage 1; blue, lineage 2; purple, lineage 3; red, lineage 4; brown, lineage 5 (M. africanum); dark green, lineage 6 (M. africanum); orange, lineage 7; light green, M. bovis.
Supplementary Figure 5 Rates of genotypic resistance to individual drugs across our entire data set of 5,310 strains.
The greatest numbers of strains were resistant to isoniazid, rifampicin and streptomycin.
Supplementary Figure 6 Overview of drug resistance mutation arisals.
The number of arisals and number of strains for each of the 392 drug resistance mutations detected in our data set.
Supplementary Figure 7 Breakdown of overall genotypic drug resistance patterns by lineage.
Data are shown for each lineage with greater than 100 strains in our data set.
Supplementary Figure 8 Breakdown of genotypic resistance to individual antituberculous drugs by predicted lineage.
M. tuberculosis strains from lineages 5, 6 and 7 were excluded because of low numbers of isolates, and lineage B was excluded as all 44 strains were fully drug susceptible.
Supplementary Figure 9 Breakdown of resistance to individual antituberculous drugs by UN geographic region.
Geographic distribution of strains in 11 UN geographic regions having greater than 30 representative isolates in this data set (45 strains that derived from 6 UN geographic regions were excluded from this figure).
Supplementary Figure 10 Breakdown of monoresistance and polyresistance by region and lineage.
(a) Breakdown of monoresistance by geographic region and by lineage. (b) Breakdown of poly-drug resistance (excluding MDR, pre-XDR and MDR) by geographic region and by lineage.
Supplementary Figure 11 Percentage of nodes where resistance to other drugs arose prior or coincident with an Xpert MTB/RIF-detectable mutation.
(a) Resistance to single drugs. (b) Resistance to multiple drugs. This figure shows results for cases where all nodes between the first and last drug acquisition event leading up to the Xpert MTB/RIF-detectable mutation fit our filtering criteria (>90% bootstrap in the more ancestral node of a pair; maximal branch lengths between pairs <1 × 10–4).
Supplementary information
Supplementary Text and Figures
Supplementary Figures 1–11, Supplementary Tables 1, 3, 4, 6 and 8–13, and Supplementary Note (PDF 3734 kb)
Supplementary Table 2
List of 5,310 strains included in our final data set of sequenced M. tuberculosis clinical isolates. (XLSX 414 kb)
Supplementary Table 5
List of all 392 mutations in our data set and their frequencies, as well as their frequencies of occurring first. (XLSX 122 kb)
Supplementary Table 7
The number of arisals and the number of strains with each mutation, for each of the 11 geographical regions with >30 strains. (XLSX 127 kb)
Rights and permissions
About this article

Cite this article
Manson, A., Cohen, K., Abeel, T. et al. Genomic analysis of globally diverse Mycobacterium tuberculosis strains provides insights into the emergence and spread of multidrug resistance. Nat Genet 49, 395–402 (2017). https://doi.org/10.1038/ng.3767
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ng.3767
This article is cited by
-
Detection of a historic reservoir of bedaquiline/clofazimine resistance-associated variants in Mycobacterium tuberculosis
Genome Medicine (2024)
-
Multidrug-resistant tuberculosis
Nature Reviews Disease Primers (2024)
-
Drug-resistant tuberculosis: a persistent global health concern
Nature Reviews Microbiology (2024)
-
Molecular Detection of Multidrug Resistance and Characterizations of Mutations in Mycobacterium Tuberculosis Using Polycarbonate Track-Etched Membrane Based DNA Bio-Chip
Indian Journal of Microbiology (2024)
-
Resistance patterns among drug-resistant tuberculosis patients and trends-over-time analysis of national surveillance data in Gabon, Central Africa
Infection (2023)

{kind=link}