
Abstract
Gene drive systems that enable super-Mendelian inheritance of a transgene have the potential to modify insect populations over a timeframe of a few years. We describe CRISPR-Cas9 endonuclease constructs that function as gene drive systems in Anopheles gambiae, the main vector for malaria. We identified three genes (AGAP005958, AGAP011377 and AGAP007280) that confer a recessive female-sterility phenotype upon disruption, and inserted into each locus CRISPR-Cas9 gene drive constructs designed to target and edit each gene. For each targeted locus we observed a strong gene drive at the molecular level, with transmission rates to progeny of 91.4 to 99.6%. Population modeling and cage experiments indicate that a CRISPR-Cas9 construct targeting one of these loci, AGAP007280, meets the minimum requirement for a gene drive targeting female reproduction in an insect population. These findings could expedite the development of gene drives to suppress mosquito populations to levels that do not support malaria transmission.
Similar content being viewed by others

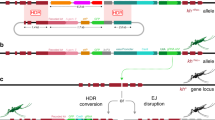
Main
Synthetic gene drive systems using site-specific endonucleases to spread traits into a population were first proposed more than a decade ago1. This proposal was initially inspired by the action of a class of natural selfish genetic elements, found in many single-cell organisms, named homing endonuclease genes (HEGs). HEG-encoded proteins can recognize and cleave a 15- to 30-bp DNA sequence. HEGs are located within the DNA recognition sequence, rendering it resistant to further cleavage. However, when the HEG comes into contact with a chromosome containing the uninterrupted recognition sequence, the double-strand break (DSB) induced by the cleavage is often repaired using the homologous chromosome as a template, effectively converting a heterozygote into a homozygote in a process known as 'homing'. Through this mechanism, the frequency of an HEG can rapidly increase in a population. Naturally occurring HEGs can in principle be adapted to function as a gene drive system in mosquitoes because they can be re-engineered to recognize mosquito genes2. An HEG expressed in the male mosquito germline that recognizes an artificially introduced recognition site shows high rates of super-Mendelian inheritance and rapidly invades a caged population2. The increased transmission rate provided by endonuclease-based gene drive systems could theoretically outweigh the fitness costs arising from the cleavage activity and disruption of the targeted sites. If this proviso is met, a drive construct can spread through a population until it reaches an equilibrium frequency, with a reduced mean fitness for the population1.
Any nuclease with a sufficiently long recognition sequence could hypothetically be redesigned to function as a gene drive system akin to an HEG, provided that it can be engineered to recognize and insert in a specific genomic locus. For example, we have previously shown that modular nucleases such as zinc finger nucleases or transcription activator–like effector nucleases (TALENs), for which the DNA-binding specificity of each module is well-characterized, can be combined to function as a synthetic selfish element in Drosophila, albeit with low replication fidelity owing to their repetitive nature3. More recently, the development of the CRISPR-Cas9 (clustered, regularly interspaced, short palindromic repeats (CRISPR) and CRISPR-associated protein (Cas)) system4,5,6 has radically simplified the process of engineering nucleases that can cleave specific genomic sequences. A guide RNA (gRNA) complementary to a DNA target site directs the activity of the Cas9 endonuclease to that sequence, providing a means to edit almost any chosen DNA sequence without the need to undertake complex protein engineering and selection procedures. In addition to applications in genome editing, the specificity and the flexibility of the CRISPR-Cas9 system offers unprecedented opportunities to expedite the development of gene drive systems for the control of insect vectors of disease7. In a proof-of-principle experiment for such a use, a CRISPR-based construct was used to demonstrate gene drive activity in a single generation at an eye color locus in Drosophila and using a split-drive system in yeast8,9.
Translation of this technology for the control of the insect vector of human malaria requires development of an endonuclease-based gene drive system that interferes with the ability of A. gambiae mosquitoes to transmit the disease. This could be achieved either by blocking parasite development or by reducing the reproductive capability of the insect vector. Modeling of vector populations indicates that the latter might be achieved through the use of an endonuclease designed to 'home' to and yield a recessive mutation in a gene that is essential for viability or female fertility, with the latter being more effective, provided that homing is temporally and spatially confined to the germline during, or before, the process of gamete formation1,10. This is essential to avoid somatic disruption of the wild-type (WT) allele and allow the normal development of heterozygous mosquitoes, critical for transmitting the endonuclease to subsequent generations. Along these lines we have developed a CRISPR-based gene drive system designed to home, in both sexes of the human malaria vector A. gambiae, to haplosufficient, somatically expressed female-fertility genes.
To identify putative female-fertility genes in A. gambiae, we used a combination of orthology and a sterility index based on a logistic regression model that correlated gene expression features with the likelihood of female sterile alleles in the model dipteran Drosophila –melanogaster11,12. Three candidate genes with high ovary expression and tissue specificity were chosen from this analysis: AGAP005958 (ortholog of Drosophila yellow-g, a haplosufficient female-fertility gene expressed in somatic follicle cells13); AGAP007280 (ortholog of Drosophila nudel, a haplosufficient female-fertility gene expressed in somatic follicle cells involved in dorsoventral patterning of the embryo14); AGAP011377 (no apparent Drosophila ortholog but contains a probable chitin binding domain).
We used either CRISPR-Cas9 nuclease or TALENs to selectively disrupt the coding sequence of these candidate genes and analyzed reproductive phenotypes to validate the suitability of these genes as homing targets. The gene knockout strategy generated 'docking lines' through homologous recombination inserting a GFP transcription unit flanked by two attP sites suitable for subsequent insertion of active drive –constructs (Fig. 1a). Though not strictly necessary for the purpose of inserting a gene drive element, the generation of the docking lines allows an unambiguous assessment of the phenotype caused by gene disruption, in the absence of ongoing Cas9 activity, and the tracking of mutant alleles by the presence of the fluorescent marker. In each case the insertion of the attP-GFP docking cassette was designed to produce a null phenotype. Both TALENs and Cas9 nuclease were effective in cutting the corresponding target sequences and in promoting the insertion of the docking construct at the cleavage site. At each of the three selected target loci, transformed GFP+ individuals were recovered at a relatively high frequency with rates at least comparable to those in our experience of transposon-mediated germline transformation (Supplementary Table 1), and they were confirmed in PCR experiments to carry the desired homologous recombination events (Supplementary Fig. 1). G1 individuals of the docking lines were fertile and were intercrossed to produce G2 progeny, expected to include individuals both heterozygous and homozygous for the insertion. Visual inspection of G2 progeny identified two classes of mosquitoes on the basis of GFP intensity, 'intermediate' and 'strong', which we attributed to the presence of one or two copies of the GFP gene in the heterozygous or homozygous state, respectively, and later confirmed by molecular analysis (Fig. 1b). Fertility assays (egg laying and hatching) performed on individual mosquitoes showed that all homozygous female mosquitoes were sterile, whereas heterozygous females showed normal rates of egg laying and hatching (Fig. 1c). On the basis of these results we concluded that the selected genes should be regarded as haplosufficient female-sterility genes. Manifestation of the impaired fertility phenotype differed across the three genes targeted, consistent with their function at distinct stages of egg production and embryo development: homozygous females carrying two disrupted alleles of either AGAP005958 or AGAP011377 failed to lay eggs, whereas homozygous mutant females at AGAP007280 laid eggs that did not hatch (Supplementary Fig. 2).

(a) A plasmid-based source of either a TALEN or Cas9 coupled with a gRNA induces a DSB at the target locus. A plasmid (hdrGFP) containing regions of homology immediately upstream and downstream of the cut site acts as a template for homology-directed repair. Internal to the homology regions a 3xP3::GFP cassette identifies hdrGFP integration events and two attP sites facilitate secondary modification of the locus through RMCE. (b) PCR was used to confirm the targeted loci in WT individuals as well as those homozygous and heterozygous for the hdrGFP allele. The primer pair used is indicated in a (blue arrows). (c) Counts of larval progeny from individual females homozygous or heterozygous for hdrGFP alleles mated to WT males. Heterozygous docking lines for all three loci showed at least full fertility compared to WT females. A minimum of 20 individuals were tested for each line. Vertical bars represent the mean and error bars the s.e.m.
After validating the female-fertility phenotype of the target genes, we inserted a gene drive construct (CRISPR homing allele, CRISPRh) (Fig. 2a) into the docking site by recombinase-mediated cassette exchange (RMCE)15. Each drive construct was designed to home, in both sexes, into the cognate WT locus and contained the following components: (i) the Cas9 nuclease gene under the control of the vasa2 promoter, shown in a previous report to be active in the germline of both sexes16; (ii) a gRNA sequence designed to direct the cleavage activity of the nuclease to the same sequence targeted in the gene-knockout experiments and under the promoter of the ubiquitously expressed, PolIII-transcribed U6 gene17; and (iii) a visual marker (3xP3::RFP). AttB sites flanking the CRISPRh construct were used to direct ΦC31 integrase-mediated recombination at the docking site. Aware of the potential for these mosquitoes to show gene drive activity, we housed our mosquitoes in a containment facility consistent with recent recommendations for safeguards in such experiments18. Successful cassette exchange events were visually identified among G1 progeny as GFP+ to RFP+ phenotype conversions, and confirmed using PCR (Supplementary Fig. 3). At all three female-fertility loci we recovered double-crossover events that resulted in cassette exchange and insertion of the CRISPRh allele, at transformation frequencies of 2–7% (Table 1). In the cassette exchange reaction we observed the insertion of both complete and incomplete CRISPRh alleles, the latter probably the result of intramolecular recombination between regions of homology in the gRNA construct and its endogenous target at the insertion site.

(a) RMCE was used to replace the GFP transcription unit in hdrGFP docking lines with a CRISPR homing construct (CRISPRh consisting of a 3xP3::RFP marker, Cas9 under the transcriptional control of the vasa2 promoter and a gRNA under the control of the ubiquitous U6 PolIII. The gRNA cleaves at the nondisrupted WT allele. Repair of the cleaved chromosome through HDR leads to copying of the CRISPRh allele and homing. (b) Confinement of homing to the germline should lead to super-Mendelian inheritance of a homing construct (indicated in red) that, when targeting a haplosufficient, somatic female-fertility gene, will reduce the number of fertile females. (c) High levels of homing at all three female-fertility loci were observed. Male or female CRISPRh/+ heterozygotes were mated to WT. Progeny from individual heterozygous females were scored for the presence of the RFP linked to the CRISPRh construct and the average transmission rate indicated by vertical bar (± s.e.m.). A minimum of 34 females were analyzed for each cross. The average homing rate is also shown. (d,e) Counts of eggs and hatching larvae for the individual crosses revealed a strong fertility effect in heterozygous CRISPRh/+ females (d) that was not seen in equivalent heterozygous males (e). (f) Dynamics calculated using recurrence equations in Deredec et al.10, using the observed homing rates in males and females and effects on female fertility. We assume no fitness effects in males and that the initial release consists of heterozygous males equal to 10% of the prerelease adult male population (i.e., 5% of the overall population). The model assumes discrete generations (one per month) and random mating; results are plotted starting from the first generation after release and do not account for evolution of either the CRISPR allele or the target sequence. (g) Increase in frequency of CRISPRh allele in cage population experiments. An equal number of CRISPRh/+ and WT individuals were used to start a population, and the frequency of individuals containing a CRISPRh allele was recorded in each subsequent generation. Black line shows deterministic prediction based on observed parameter values (homing rates 98.4%, heterozygous female fitness of 9.3%, homozygous females completely sterile), assuming no fitness effects in males. Gray lines show results from 20 stochastic simulations assuming 300 males and 300 females are used to start the next generation, females mate randomly with a single male and 15% of females fail to mate, using random numbers drawn from the appropriate multinomial distributions. Red lines show results from two replicate cages.
Each complete integration event generated a CRISPRh allele encoding a Cas9-gRNA endonuclease designed to target the corresponding integration site on a WT chromosome. Accordingly, the CRISPRh allele was resistant to nuclease cleavage as its target sequence had been interrupted by the insertion of the CRISPRh construct itself. In heterozygous mosquitoes the activation of the vasa2 promoter during gamete formation should induce the synthesis of the Cas9 nuclease that, in concert with the ubiquitously expressed gRNA, should cleave the target sequence in the fertility genes, thereby initiating homologous recombination repair events that lead to the homing of the CRISPRh construct into the WT allele (Fig. 2a). Visual screening was used to analyze the frequency of the RFP-linked CRISPRh allele in the progeny of heterozygous parents crossed to WT mosquitoes to detect signs of non-Mendelian inheritance, above the expected frequency of 50%, that would reveal gene drive activity (Fig. 2b).
In several of the CRISPRh/+ G1 individuals that we recovered at each locus we noticed super-Mendelian inheritance of the RFP-marked CRISPRh allele, with rates of 94.4–100% (Table 1) among the progeny. To further investigate the activity of these CRISPRh alleles, we looked at homing ability and sterility in the G2 generation and beyond, scoring the progeny of large numbers of single crosses to WT mosquitoes. Invariably, we saw high rates of transmission in every fertile cross we examined (Fig. 2c), representing average homing rates (defined as the proportion of non-CRISPR alleles converted to CRISPRh in the gametes) ranging from 87.3% to 99.3% across the three target genes. Importantly, though we observed more variability (69–98%) across generations over time, we observed no obvious decrease in homing performance (Table 2), suggesting that the majority of CRISPR homing events regenerate an intact allele. Furthermore, the transmission rate of the CRISPRh allele at AGAP007280 and AGAP011377 was high in both male and female CRISPRh/+ individuals, in agreement with the predicted activity of the vasa2 promoter in both sexes during early gametogenesis16. In those rare progeny that did not contain a CRISPR homing allele, we looked for evidence of repair by nonhomologous end joining (NHEJ), microhomology-mediated end joining (MMEJ)19 or other noncanonical homing events at the three target loci. In a total of 32 offspring derived from a minimum of 7 individuals, we found a total of 13 indel mutations (6 unique, including two examples of a 6-bp deletion that preserved reading frame and could represent a resistant allele), presumably arising from NHEJ or MMEJ repair, and two events from the same parent producing a 195-bp insertion at AGAP007280, most parsimoniously explained by an incomplete homing event that was resolved using homology between the gRNA sequence in the construct and its cognate target in the genome (Supplementary Fig. 4). Consistent with rare incomplete homing events generating a nonfunctional homing allele, we recovered an identical event in a single individual that produced progeny with a normal Mendelian segregation of the transgenic phenotype.
Though homing rates were high in the germline of both males and females, the fertility of females heterozygous for a homing construct was markedly reduced, with the number of larvae produced only 4.6% of WT (bootstrap 95% confidence limits 2.3–7.7%) for AGAP011377 and 9.3% (5.7–14.2%) of WT for AGAP007280. We did not recover a single larva from females heterozygous for a CRISPRh allele at AGAP005958 (Fig. 2d). In contrast, males heterozygous for CRISPRh alleles showed normal fertility (Fig. 2e). The fertility reduction observed for heterozygous CRISPRh females was at odds with the phenotype observed in heterozygous docking line females where the disruption of single alleles of AGAP011377, AGAP007280 and AGAP005958 apparently did not affect female fertility. This reduction in fertility is probably due to somatic expression of the Cas9 nuclease, as we have observed for a similar construct targeting GFP (Supplementary Fig. 5), and as others have observed in Drosophila9,20. The nos promoter has recently been found to be substantially more germline-specific in directing Cas9 activity in Drosophila20, and our system is flexible so it can accommodate alternative promoters.
Our measures of homing rates and fertility effects can be used with the model of Deredec et al.10 to derive an initial prediction about whether the constructs would be expected to spread if released into a population. This analysis revealed that the fitness cost in terms of reduced reproductive capability imposed by the CRISPRh constructs at AGAP011377 and AGAP005958 outweigh the homing rate, and the constructs would be expected to disappear from a population over time—in many aspects these constructs match the requirements of female-specific RIDL (release of insects with a dominant lethal) with enhanced transmission21, a potent form of the sterile insect technique, though conditional rescue of the sterility may be required for efficient production. However, the higher homing rates observed for CRISPRh at AGAP007280, combined with the milder fertility reduction observed in heterozygous females indicate that this construct could spread through a population, at least initially, and impose a reproductive load on the population as it does so, fulfilling one of the major requirements for a functional gene drive measure for vector control (Fig. 2f). To investigate the ability of the CRISPRh allele to spread at the AGAP007280 locus, caged populations were initiated with CRISPRh/+ and WT individuals at equal frequency and monitored over several generations. Consistent with the modeling predictions we observed a progressive increase in the frequency of individuals positive for the CRISPRh allele from 50% to 75.1% over four generations (Fig. 2g). Such a reproductive load will impose a strong selection pressure for resistant alleles, some of which will be generated by the gene drive system itself through NHEJ or MMEJ repair of endonuclease-induced chromosome breaks, as we previously showed molecularly (Supplementary Fig. 4). The longer term dynamics will depend on the efficiency of spreading on the one hand and the fitness cost of mutations arising at the cleavage site on the other hand10,22. Ultimately the effect of these mutations could be mitigated by designing nucleases that target conserved, functionally constrained regions in the target gene and that are tolerant of mutations1. This could be achieved using a CRISPR-Cas9 gene drive through the use of multiple gRNAs targeting sequence variants7.
The high frequency with which gene knockouts were achieved at three separate loci and the ease with which these could be both tracked using a visual marker and secondarily modified to include genes of choice verify the CRISPR-Cas9 gene drive system as a robust gene editing tool that will be valuable for functional genetics in the malaria mosquito. The rates of super-Mendelian inheritance that we observed with CRISPR-based homing constructs at female-fertility loci establish a solid basis for the development of a gene drive system that has the potential to substantially reduce mosquito populations. Moreover, our gene drive element was able to carry substantial additional sequence in the form of the RFP marker unit, indicating that this technology is also resilient to bringing along additional cargo, making it suitable for population-modification strategies that are aimed at modifying vector populations with transgenes conferring useful phenotypes such as parasite resistance. Being able to use CRISPR-Cas9 in mosquitoes means that genome editing and nuclease engineering will no longer be technical bottlenecks in this major pest insect.
The success of gene drive technology for vector control will depend on the choice of suitable promoters to effectively drive homing during the process of gametogenesis, the phenotype of the disrupted genes, the robustness of the nuclease during homing and the ability of the target population to generate compensatory mutations.
Methods
Choice of target genes.
Sterility Index – p(sterile).
To assess the likely effects on sterility as a result of gene inactivation, we created a sterility index with logistic regression models in Drosophila on the basis of gene expression and the correlated effects of genetic knock before applying model parameters to the Anopheles genome. The models were analyzed with the R statistical programming language (http://www.r-project.org/).
Gene expression.
MozAtlas11 and FlyAtlas23 gene expression estimates were obtained for both Anopheles and Drosophila probe sets. In order to make Anopheles gene expression comparable with Drosophila, we pooled together sex-specific samples and recorded the maximum intensity in either sex. If multiple probes were present for a gene, expression in each tissue was calculated as maximum probe intensity, whereas probes present in multiple genes were omitted from further analysis. Only probes indicating expression as 'present' in at least three of four biological replicates were included in this analysis. Models were constructed on the basis of rank normalized gene expression in the Head, Carcass, Testis and Ovary of Drosophila gene expression. For each tissue, gene expression was ranked from lowest to highest expression intensity (ties were allocated the minimum rank for that group of genes) and divided by the number of genes in the data set. Rank-normalized values fall between 0 and 1, which reflect the proportion of genes with a lower expression value in that tissue, for example, a value of 0.8 indicates that 80% of other genes in that tissue have a lower expression level. Tissue specificity was represented by the tau-statistic24. Expression was normalized in each tissue against maximum expression for that gene. These values are divided by the number of tissues (n – 1) and subsequently summed together. The value will lie between 0 and 1. A value of 1 equals specific expression. A value of 0 is equal to ubiquitous expression.
Phenotype annotations.
Phenotype annotations were obtained from FlyBase (2011_7) (ref. 25) (Supplementary Table 2). For modeling, annotatio of a null sterile annotation. Specifically, genes with more than ten alleles, but not annotated as sterile, were included in the model as NONSTERILE (n = 1,509). Genes annotated with a sterile identifier, and either more than ten alleles or evidence of a null sterile mutation were included in models as STERILE (n = 536). The remaining genes were left as UNKNOWN and not included in modeling (n = 8,886).
Logistic regression.
The results of the logistic regression are shown in Supplementary Table 3. The product (ovary:tau) of ranked ovary expression and tissue specificity had the highest correlation with a female sterile annotation. Once we extended the coefficients obtained for Drosophila to the Anopheles expression data set for the same tissues, we found 271 Anopheles genes with a P(sterile) score ≥0.5 (the three genes chosen for this study are shown in Supplementary Table 4, the full list is provided in Supplementary Table 5). We refer to the P(sterile) value of genes as the sterility index.
Generation of donor constructs for gene targeting by CRISPR or TALEN-mediated HDR.
Gene-targeting vectors were assembled by Gateway cloning (Invitrogen) and designed to contain an attP-flanked 3xP3::GFP marker construct enclosed within homology arms extending 2 kb either direction of the expected CRISPRh cleavage site, as well as an external 3xP3::RFP marker. Regions flanking the target sites for each gene were amplified with primers that included the necessary recombinase sites for the Gateway reaction (underlined). For AGAP005958: 5958-T1[5′F1]B1 (GGGGACAAGTTTGTACAAAAAAGCAGGCTGTGCAAGCTAGCCGTTTCGAG) and 5958-T1[5′R1]B4 (GGGGACAACTTTGTATAGAAAAGTTGGGTGCGCGGCTCCAGTATCTCGTCA) as well as 5958-T1[3′F1]B3 (GGGGACAACTTTGTATAATAAAGTTGAGCTGGATTTCACAATCTCCGA) and 5958-T1[3′R1]B2 (GGGGACCACTTTGTACAAGAAAGCTGGGTACTCGTGCATTTGACTGCTTCC) to generate the left and right arms of homology, respectively. Regions flanking the AGAP007280 target site were amplified using 7280-T1[5′F1]B1 (GGGGACAAGTTTGTACAAAAAAGCAGGCTCAGATACTGATGCCGCAGGTTCA) and 7280-T1[5′R1]B4 (GGGGACAACTTTGTATAGAAAAGTTGGGTGGAAAGTGAGGAGGAGGGTGGTAGTG) as well as 7280-T1[3′F1]B3 (GGGGACAACTTTGTATAATAAAGTTGTTTCTTCCTCACCTCGCTGCGA) and 7280-T1[3′R1]B2 (GGGGACCACTTTGTACAAGAAAGCTGGGTACCCCTCCAGCTATGATCAACATGC) to generate the left and right arms of homology, respectively. Regions flanking the AGAP011377 target site were amplified using 11377-T1[5′F1]B1 (GGGGACAAGTTTGTACAAAAAAGCAGGCTCTAGTGGCTACAGGCAGGCC) and 11377-T1[5′R1]B4 (GGGGACAACTTTGTATAGAAAAGTTGGGTGGAAATTTTCCGGCGCCAGGC) as well as 11377-T1[3′F1]B3 (GGGGACAACTTTGTATAATAAAGTTGTTTCTACGTCTGCTACAACG) and 11377-T1[3′R1]B2 (GGGGACCACTTTGTACAAGAAAGCTGGGTAGACGAGTCAACTCCAGGGCT) to generate the left and right arms of homology, respectively.
The amplified left and right homology arms were cloned by BP reaction (Invitrogen) into pDONR221-P1P4 and pDONR221-P3P2, respectively. The resultant pENTR vectors were assembled into donor vectors (pHDRgfp-11377; pHDRgfp-5958; pHDRgfp-7280) by LR reaction (Invitrogen) with an attP-GFP-attP pENTR vector and a destination vector containing a 3xP3::RFP marker external to the arms of the homology that should not be inserted into the target locus during a legitimate homology-directed repair event.
Generation of CRISPR and TALEN constructs.
A human codon-optimized version of the Streptococcus pyogenes Cas9 gene (hCas9) was amplified from pX330 (AddGene/Zhang laboratory) using primers containing SalI and PacI sites, SalI-hCas9-F (aacgtcgacGATCCCGGTGCCACCATGGA) and PacI-hCas9-R (aacttaattaaTTTCGTGGCCGCCGGCCTTTT). hCas9 was then subcloned with SalI and PacI into a vasa2 promoter–containing vector before cloning into a RMCE vector synthesized by DNA2.0 to contain the vasa 3′ UTR regulatory sequence and a U6::gRNA cassette containing a spacer cloning site based on Hwang et al.26, all flanked by attB recombination sites. The U6 snRNA polymerase III promoter and terminator sequences were used as described previously17. The resultant vector, p165, was digested with BsaI and modified to contain individual gRNA spacers by Golden Gate cloning of appropriately designed and annealed oligos bearing complete homology to the intended target sequence with unidirectional overhangs compatible with BsaI-digested p165. The full sequence of vector p165 has been deposited to GenBank (accession ID: KU189142). The resultant vectors containing gRNAs targeting AGAP011377 (gRNA sequence: GCAGACGTAGAAATTTTC), AGAP005958 (GAGATACTGGAGCCGCGAGC) and AGAP007280 (GGAAGAAAGTGAGGAGGA) were named p16503, p16505 and p16501 respectively. Individual gRNA target sites were identified and assessed for off-targets using both the ZiFiT (http://zifit.partners.org/) and ChopChop (https://chopchop.rc.fas.harvard.edu) websites.
TALEN binding sites targeting the AGAP011377 gene were selected using the TALE-NT software27 and the site TCGAAAACACGGGCctggcgccggaaaatTTCTACGTCTGCTAC was chosen to cleave at AGAP011377 at a site that overlapped with the corresponding CRISPR site (underlined) in the same gene (Supplementary Fig. 6). TALEN-expressing plasmids were assembled by Golden Gate cloning as described8 using the GoldyTALEN scaffold as destination vector28. Subsequently, each TALEN monomer was cloned into an Anopheles expression vector under the expression of the vasa2 promoter and 3′UTR and the FoKI cleavage domains were modified to be active as obligate heterodimer (DD/RR variants).
The location of the TALEN and CRISPR recognition sites in relation to the coding sequence of the target genes is shown in Supplementary Figure 6. Each recognition sequence is followed by the obligatory PAM sequence of 5′-NGG distal to the region of complementarity in the gRNA sequence.
Microinjection of mosquito embryos and selection of transformants.
Freshly laid embryos of the Anopheles gambiae G3 strain, herein referred to as wild type, reared under standard conditions of 70% relative humidity at 26 ± 2 °C were used for microinjections as described elsewhere29.
For the generation of the hdrGFP docking lines the donor construct (300 ng/μl) containing regions of homology to the relevant target locus was injected together with the relevant CRISPR plasmid (300 ng/μμl) for AGAP007280 (p16501) and AGAP005958 (p16505) or, for AGAP011377, plasmids expressing the left and right monomers of the TALEN (each at 300 ng/μl). Surviving G0 individuals were crossed to WT and positive transformants were identified under fluorescence microscopy as GFP+ larvae among the G1 progeny.
For the recombinase-mediated cassette exchange reactions a mix containing the relevant CRISPR plasmid (200 ng/μl) and 400 ng/μl vasa2::integrase helper plasmid30 was injected into embryos of the hdrGFP docking lines. Progeny from the outcross of surviving G0 individuals to WT were screened for the presence of RFP and the absence of GFP that should be indicative of a successful cassette-exchange event.
Containment of mosquitoes transformed with a gene drive.
All mosquitoes were housed at Imperial College London in an insectary that is compliant with Arthropod Containment Guidelines Level 2 (ref. 31). All work with transformed mosquitoes was performed under institutionally approved biosafety and protocols for genetically modified organisms. In particular GM mosquitoes containing constructs with the potential to show gene drive activity were housed in dedicated cubicles, separated by at least six doors from the external environment and requiring two levels of security card access. Moreover, because of its location in a city with a Northern temperate climate, Anopheles gambiae mosquitoes housed in the insectary are also ecologically contained. The physical and ecological containment of the insectary are compliant with guidelines set out in a recent commentary calling for safeguards in the study of synthetic gene drive technologies18.
Molecular confirmation of gene targeting and recombinase-mediated cassette exchange.
To confirm molecularly the successful integration of docking or CRISPRh constructs into their genomic target site, genomic DNA was extracted using the Wizard Genomic DNA purification kit (Promega) from GFP+ or RFP+ G1 mosquitoes respectively. Docking sites were interrogated by PCR using primers binding the docking construct, 5′GFP-R (TGAACAGCTCCTCGCCCTTG) and 3′GFP-F (GCCCTGAGCAAAGACCCCAA) with primers binding the genome outside of the homology arms (as portrayed in Supplementary Fig. 1): AGAP11377 using DL-11377-F (GGGTGTTAACGTTCCGCCTA) and DL-11377-R (ACCCAAGACCACCCAAAGAC), AGAP005958 using DL-5958-F1 (CGGACACGCGGAAGTCTGAA) and DL-5958-R1 (CGACTTTCCCGGAACATTTACCA), and AGAP007280 using DL-7280-F1 (AGCACGTGCCGGCTAAAGCT) and DL-7280-R1 (GCCACACCAGCAACAGCCTTATC). For the purposes of producing a shorter amplicon that could reliably amplify both the WT allele and the hdrGFP allele in the same PCR reaction (e.g., Fig. 1), the following primer pairs were used: AGAP011377 (Seq-11377-F (AACCGACAGTCCATCCTTGT) and Seq-11377-R (GAGCGTCTTTCGACCTGTTC)); AGAP007280 (Seq-7280-F (GCACAAATCCGATCGTGACA) and –Seq-7280-R (CAGTGGCAGTTCCGTAGAGA)); AGAP005958 –(Seq-5958-F (GCACTCGTCCGCGTTCTGAA) and Seq-5958-R (TTTGTGCTGGTGTCCGCGCT)).
Successful cassette exchange of CRISPRh alleles was interrogated by PCR using primers binding the CRISPRh construct, RFP2q-F (GTGCTGAAGGGCGAGATCCACA) and hCas9-F7 (CGGCGAACTGCAGAAGGGAA) with primers binding the genome: AGAP011377 using Seq-5958-F and Seq-5958-R, AGAP005958 using Seq-5958-F and Seq-5958-R, and AGAP007280 using Seq-7280-F and Seq-7280-R.
Molecular confirmation of CRISPR activity at target loci.
To assess molecularly the activity of CRISPR at the target locus, the target site was sequenced in those progeny (RFP–) that apparently failed to receive a CRISPR homing allele from a hemizygous RFP+ parent. Genomic DNA was extracted using the Wizard Genomic DNA purification kit (Promega). Amplicons 2.5 kb either side of the CRISPRh target site in AGAP011377, AGAP005958 and AGAP007280 were amplified with Phusion HF polymerase (Thermo Scientific) using Seq-11377-F and Seq-11377-R, Seq-5958-F and Seq-5958-R, and Seq-7280-F and Seq-7280-R primers (described above), respectively. PCR products were purified (Qiagen PCR purification kit) and sequenced using internal primers, Seq-11377-F2 (TCGCCATGTACGCCACCAAC), Seq-5958-F2 (CTTGCCGCTGCGCAGATGTT) and Seq-7280-F2 (TCCGGTGGACCGTTTGTGTG). The sequences of the target loci (both WT and those showing mutations) have been deposited in GenBank (accession IDs: KU183683-KU183700).
Fertility assays.
Heterozygous 'docking line' individuals were intercrossed to generate heterozygous and homozygous hdrGFP mutants. Offspring were screened for homozygous or heterozygous knock-in mutations by 'strong' or 'intermediate' intensity of GFP fluorescence, respectively. Male and female individuals from each screened homozygous and heterozygous class were mated to an equal number of wild-type mosquitoes for 5 d. Females were blood fed on an anesthetized mouse on the sixth day and a minimum of 40 mosquitoes were isolated individually into 300-ml beakers and allowed to lay 3 d later into a 25-ml cup filled with water and lined with filter paper32. For each female, eggs and larvae were counted and 16 larvae were screened for GFP expression using a Nikon inverted fluorescence microscope (Eclipse TE200) to confirm parental hetero/homozygosity at the HDR locus. Heterozygotes were confirmed by the presence of GFP− progeny. Females that did not give larvae were dissected and checked under a microscope for the presence of sperm in their spermathecae. Those which were unmated were excluded from the phenotypic analysis.
To confirm HDR hetero/homozygosity in transgenic females that were mated but failed to give progeny, a PCR was performed across the HDR locus using a primer pair designed to amplify both the WT allele (1 kb) and the HDR+ allele (2.5 kb). The following primer pairs were used for the three genes: AGAP011377 using Seq-11377-F and Seq-11377-R), AGAP005958 using Seq-5958-F and Seq-5958-R, and AGAP007280 using Seq-7280-F and Seq-7280-R.
Fertility assays were performed using CRISPRh hemizygotes essentially the same as the docking line phenotype assays with the exception that 50 progeny from each parent were screened for the presence of an RFP-linked CRISPRh allele to assess the frequency of CRISPRh transmission.
Ethics statement.
All animal work was conducted according to UK Home Office Regulations and approved under Home Office License PPL 70/6453.
Cage experiments.
First instar mosquito larvae heterozygous for the CRISPRh allele at AGAP007280 were mixed within 12 h of eclosion with an equal number of age-matched WT larvae in rearing trays at a density of 200 per tray (in ∼1 liter rearing water). The mixed population was used to seed two starting cages with 600 adult mosquitoes each. For four generations, each cage was fed after allowing 5 d for mating, and an egg bowl placed in the cage 48 h after a blood meal to allow overnight oviposition. After allowing full eclosion, a random sample of offspring were scored under fluorescence microscopy for the presence or absence of the RFP-linked CRISPRh allele, then reared together in the same trays and 600 were used to populate the next generation.
Accession codes.
GenBank: KU183683–KU183700 (sequences of the target loci, both wild type and those showing mutations); KU189142 (full sequence of vector p165).
References
Burt, A. Site-specific selfish genes as tools for the control and genetic engineering of natural populations. Proc. R. Soc. Lond. B 270, 921–928 (2003).
Windbichler, N. et al. A synthetic homing endonuclease-based gene drive system in the human malaria mosquito. Nature 473, 212–215 (2011).
Simoni, A. et al. Development of synthetic selfish elements based on modular nucleases in Drosophila melanogaster. Nucleic Acids Res. 42, 7461–7472 (2014).
Fu, Y., Sander, J.D., Reyon, D., Cascio, V.M. & Joung, J.K. Improving CRISPR-Cas nuclease specificity using truncated guide RNAs. Nat. Biotechnol. 32, 279–284 (2014).
Hsu, P.D. et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol. 31, 827–832 (2013).
Jinek, M. et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816–821 (2012).
Esvelt, K.M., Smidler, A.L., Catteruccia, F. & Church, G.M. Concerning RNA-guided gene drives for the alteration of wild populations. eLife e03401 (2014).
DiCarlo, J.E., Chavez, A., Dietz, S.L., Esvelt, K.M. & Church, G.M. Safeguarding CRISPR-Cas9 gene drives in yeast. Nat. Biotechnol. doi:10.1038/nbt.3412 (16 November 2015).
Gantz, V.M. & Bier, E. Genome editing. The mutagenic chain reaction: a method for converting heterozygous to homozygous mutations. Science 348, 442–444 (2015).
Deredec, A., Burt, A. & Godfray, H.C. The population genetics of using homing endonuclease genes in vector and pest management. Genetics 179, 2013–2026 (2008).
Baker, D.A. et al. A comprehensive gene expression atlas of sex- and tissue-specificity in the malaria vector, Anopheles gambiae. BMC Genomics 12, 296 (2011).
Giraldo-Calderón, G.I. et al. VectorBase: an updated bioinformatics resource for invertebrate vectors and other organisms related with human diseases. Nucleic Acids Res. 43, D707–D713 (2015).
Claycomb, J.M., Benasutti, M., Bosco, G., Fenger, D.D. & Orr-Weaver, T.L. Gene amplification as a developmental strategy: isolation of two developmental amplicons in Drosophila. Dev. Cell 6, 145–155 (2004).
Hong, C.C. & Hashimoto, C. The maternal nudel protein of Drosophila has two distinct roles important for embryogenesis. Genetics 143, 1653–1661 (1996).
Bateman, J.R., Lee, A.M. & Wu, C.T. Site-specific transformation of Drosophila via phiC31 integrase-mediated cassette exchange. Genetics 173, 769–777 (2006).
Papathanos, P.A., Windbichler, N., Menichelli, M., Burt, A. & Crisanti, A. The vasa regulatory region mediates germline expression and maternal transmission of proteins in the malaria mosquito Anopheles gambiae: a versatile tool for genetic control strategies. BMC Mol. Biol. 10, 65 (2009).
Konet, D.S. et al. Short-hairpin RNA expressed from polymerase III promoters mediates RNA interference in mosquito cells. Insect Mol. Biol. 16, 199–206 (2007).
Akbari, O.S. et al. BIOSAFETY. Safeguarding gene drive experiments in the laboratory. Science 349, 927–929 (2015).
Bae, S., Kweon, J., Kim, H.S. & Kim, J.S. Microhomology-based choice of Cas9 nuclease target sites. Nat. Methods 11, 705–706 (2014).
Port, F., Chen, H.M., Lee, T. & Bullock, S.L. Optimized CRISPR/Cas tools for efficient germline and somatic genome engineering in Drosophila. Proc. Natl. Acad. Sci. USA 111, E2967–E2976 (2014).
Thomas, D.D., Donnelly, C.A., Wood, R.J. & Alphey, L.S. Insect population control using a dominant, repressible, lethal genetic system. Science 287, 2474–2476 (2000).
Deredec, A., Godfray, H.C. & Burt, A. Requirements for effective malaria control with homing endonuclease genes. Proc. Natl. Acad. Sci. USA 108, E874–E880 (2011).
Chintapalli, V.R., Wang, J. & Dow, J.A. Using FlyAtlas to identify better Drosophila melanogaster models of human disease. Nat. Genet. 39, 715–720 (2007).
Yanai, I. et al. Genome-wide midrange transcription profiles reveal expression level relationships in human tissue specification. Bioinformatics 21, 650–659 (2005).
Drysdale, R.A. & Crosby, M.A. FlyBase Consortium. FlyBase: genes and gene models. Nucleic Acids Res. 33, D390–D395 (2005).
Hwang, W.Y. et al. Efficient genome editing in zebrafish using a CRISPR-Cas system. Nat. Biotechnol. 31, 227–229 (2013).
Cermak, T. et al. Efficient design and assembly of custom TALEN and other TAL effector-based constructs for DNA targeting. Nucleic Acids Res. 39, e82 (2011).
Carlson, D.F. et al. Efficient TALEN-mediated gene knockout in livestock. Proc. Natl. Acad. Sci. USA 109, 17382–17387 (2012).
Fuchs, S., Nolan, T. & Crisanti, A. Mosquito transgenic technologies to reduce Plasmodium transmission. Methods Mol. Biol. 923, 601–622 (2013).
Volohonsky, G. et al. Tools for Anopheles gambiae transgenesis. G3 (Bethesda) 5, 1151–1163 (2015).
Scott, T.W. Containment of arthropod disease vectors. ILAR J. 46, 53–61 (2005).
Thailayil, J., Magnusson, K., Godfray, H.C., Crisanti, A. & Catteruccia, F. Spermless males elicit large-scale female responses to mating in the malaria mosquito Anopheles gambiae. Proc. Natl. Acad. Sci. USA 108, 13677–13681 (2011).
Acknowledgements
We are grateful to C. Bamikole, A. Hall and D. Fabrigar for experimental assistance and to P. Papathanos and S. Fuchs for useful discussion. This work was supported by a grant from the Foundation for the National Institutes of Health through the Vector-Based Control of Transmission: Discovery Research (VCTR) program of the Grand Challenges in Global Health initiative of the Bill & Melinda Gates Foundation and by the European Research Council under the European Union's Seventh Framework Programme ERC grant no. 335724 to N.W.
Author information
Authors and Affiliations
Contributions
A.H., R.G., K.K., A.S., C.S., D.K. and M.G. performed the experiments. E.M. and N.W. developed mosquito reagents. D.B., S.R., T.N. and A.B. developed the logistic regression model. A.B. did the experimental modeling. A.H., R.G., N.W. and T.N. designed the experiments. A.H., A.C., A.B. and T.N. analyzed the data. A.H., A.C. and T.N. wrote the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Integrated supplementary information
Supplementary Figure 1 Molecular confirmation ofCRISPR–induced HDR at 3 candidate female fertility loci
A PCR was performed using a primer pair (blue arrows) specific for either the 5′ or 3′ side of the putative integration of the hdrGFP docking construct. In each case a primer binding internal to the hdrGFP construct was combined with a primer binding external to the region of homology included in the hdrGFP plasmid such that an amplicon of the expected size should only be produced in the event of the precise homol-ogy-dependent repair event illustrated. 3 individuals of each line were tested.
Supplementary Figure 2 Docking lines containing HDR gene disruptions show sterility at different stages of egg maturation.
Females homozygous and heterozygous for the hdrGFP allele were mated in groups to wild type males and al-lowed to lay individually and the number of resulting eggs and larvae counted. A minimum of 20 individuals were tested for each line.
Supplementary Figure 3 Molecular confirmation of CRISPRh alleles at candidate female fertility loci
Following RMCE with a CRISPRh construct a PCR was performed to confirm the molecular nature of the event using a primer pair (blue arrows) designed to amplify from the docking site in the respective gene (blue bar) to an internal region of the CRISPRh construct. Primer pairs were designed for both the 5′ and 3′ ends of the integration. Nature Biotechnology
Supplementary Figure 4 NHEJ and non–canonical CRISPR induced homing events at target loci
32 progeny from CRISPRh/+ parents that did not show the RFP+ phenotype associated with the CRISPRh con-struct were analysed. The sequence included in the protospacer of the guide RNA is highlighted, with the PAM region in blue. Across the 3 loci we recovered 15 instances of mutations at the target loci and 17 instances showing wild type sequences. Among the mutations we recovered, 6 unique (originating from separate parents) NHEJ events resulting in indels and one unique incomplete homing event at AGAP007280 that incorporated a small region surrounding the guide RNA in the homing construct (lower panel).
Supplementary Figure 5 Somatic activity of vasa::Cas9
Progeny from a homozygous vasa::Cas9;3XP3::YFP male crossed to a heterozygous U6::gRNA(YFP) female show variegated YFP expression in somatic tissues, including eye, dorsal ganglion and anal palps (left) as well as extensive mottling throughout the body (right). The two phenotypes segregated approximately 50:50, consis-tent with somatic expression of vasa::Cas9 in all individuals and Mendelian segregation of the gRNA allele to half the progeny. Larvae were visualised under a GFP filter.
Supplementary Figure 6 Schematic of the three target genes used in this study and the nuclease binding sequences.
The CRISPR target sequences are highlighted in grey; each includes a protospacer adjacent motif (PAM) se-quence at the target locus (5′-NGG), highlighted in light blue. The TALEN binding sites (for AGAP011377) are shown as underlined text.
Supplementary information
Supplementary Text and Figures
Supplementary Figures 1–6 (PDF 3012 kb)
Supplementary Table 1
CRISPR or TALEN-mediated HDR to disrupt candidate female fertility genes and produce docking lines (XLSX 10 kb)
Supplementary Table 2
Sterility annotations and controlled vocabulary (FlyBase 2011_7) (XLSX 10 kb)
Supplementary Table 3
Logistic regression coefficients (XLSX 9 kb)
Supplementary Table 4
P(sterile) of target genes (XLSX 10 kb)
Supplementary Table 5
List of genes with a p(sterile) score of ≥0.5 (XLSX 17 kb)
Rights and permissions
About this article

Cite this article
Hammond, A., Galizi, R., Kyrou, K. et al. A CRISPR-Cas9 gene drive system targeting female reproduction in the malaria mosquito vector Anopheles gambiae. Nat Biotechnol 34, 78–83 (2016). https://doi.org/10.1038/nbt.3439
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nbt.3439
This article is cited by
-
CRISPR-mediated germline mutagenesis for genetic sterilization of Anopheles gambiae males
Scientific Reports (2024)
-
Gene drive and genetic sex conversion in the global agricultural pest Ceratitis capitata
Nature Communications (2024)
-
A multiplexed, confinable CRISPR/Cas9 gene drive can propagate in caged Aedes aegypti populations
Nature Communications (2024)
-
Genome editing in pests: basic science to applications
Journal of Pest Science (2024)
-
Bioinformatic and literature assessment of toxicity and allergenicity of a CRISPR-Cas9 engineered gene drive to control Anopheles gambiae the mosquito vector of human malaria
Malaria Journal (2023)
