Article Text

Abstract
Background Invasive non-typhoidal Salmonella (iNTS) is one of the leading causes of bacteraemia in sub-Saharan Africa. We aimed to provide a better understanding of the genetic characteristics and transmission patterns associated with multi-drug resistant (MDR) iNTS serovars across the continent.
Methods A total of 166 iNTS isolates collected from a multi-centre surveillance in 10 African countries (2010–2014) and a fever study in Ghana (2007–2009) were genome sequenced to investigate the geographical distribution, antimicrobial genetic determinants and population structure of iNTS serotypes–genotypes. Phylogenetic analyses were conducted in the context of the existing genomic frameworks for various iNTS serovars. Population-based incidence of MDR-iNTS disease was estimated in each study site.
Results Salmonella Typhimurium sequence-type (ST) 313 and Salmonella Enteritidis ST11 were predominant, and both exhibited high frequencies of MDR; Salmonella Dublin ST10 was identified in West Africa only. Mutations in the gyrA gene (fluoroquinolone resistance) were identified in S. Enteritidis and S. Typhimurium in Ghana; an ST313 isolate carrying blaCTX-M-15 was found in Kenya. International transmission of MDR ST313 (lineage II) and MDR ST11 (West African clade) was observed between Ghana and neighbouring West African countries. The incidence of MDR-iNTS disease exceeded 100/100 000 person-years-of-observation in children aged <5 years in several West African countries.
Conclusions We identified the circulation of multiple MDR iNTS serovar STs in the sampled sub-Saharan African countries. Investment in the development and deployment of iNTS vaccines coupled with intensified antimicrobial resistance surveillance are essential to limit the impact of these pathogens in Africa.
- epidemiology
- medical microbiology
- typhoid and paratyphoid fevers
- cross-sectional survey
Data availability statement
Data are available in a public, open access repository. Raw sequence data are available in the European Nucleotide Archive (Project number: ERP009684, ERP010763, ERP013866).
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Key questions
What is already known?
Invasive non-typhoidal Salmonella (iNTS) disease is an emerging pathogen in sub-Saharan Africa.
iNTS is now a leading cause of bacteraemia in sub-Saharan Africa.
The disease is associated with specific sequence types of S. Enteritidis and S. Typhimurium.
Key questions
What are the new findings?
iNTS disease is widespread across the sampled locations.
Salmonella Typhimurium sequence-type (ST) 313 and Salmonella Enteritidis ST11 are the predominant pathogens.
There is a high frequency of multi-drug resistant (MDR) phenotypes.
The incidence of MDR-iNTS disease exceeded 100/100 000 person-years-of-observation in children aged <5 years in several West African countries.
What do the new findings imply?
Sustained disease surveillance of MDR-iNTS organisms is essential.
There is a need for acceleration of iNTS vaccines.
We need a better understanding of the disease reservoir and human-to-human transmission.
Background
The non-typhoidal members of Salmonella enterica are archetypal zoonotic pathogens typically associated with self-limiting diarrhoea in humans. However, certain non-typhoidal Salmonella serovars are also a recognised cause of invasive disease in specific geographical regions. Invasive non-typhoidal Salmonella (iNTS) is most commonly observed in infants and young adults with HIV, malaria and malnutrition.1 Annually, there are an estimated 3.4 million cases of iNTS globally, 20% of which are fatal.2 3 The vast majority of iNTS disease is reported in sub-Saharan Africa, where annual incidence rates of 175–388 cases per 100 000 person-years and case fatality rates as high as 25% have been reported in young children.4–6 A recent multi-centre study across 10 countries in sub-Saharan Africa identified iNTS as a major cause of bacteraemia in febrile patients, with incidence rates exceeding 100 cases per 100 000 person-years recorded in multiple sampling locations.7
Various Salmonella serovars have been associated with iNTS disease, including S. Isangi,8 S. Concord,9 S. Stanleyville and S. Dublin.10 However, the majority of iNTS infections in sub-Saharan Africa can be attributed to S. Typhimurium and S. Enteritidis.11 12 Salmonella Typhimurium iNTS in sub-Saharan Africa is largely associated with a multi-drug resistant (MDR; resistant to ampicillin, chloramphenicol and trimethoprim–sulfamethoxazole) variant of ST313, which comprised two major lineages (I and II).11 Salmonella Enteritidis account for approximately one-third of the iNTS cases reported in sub-Saharan Africa and are primarily associated with genotype ST11. There are three major clades of S. Enteritidis ST11 (Global epidemic, West African and Central/East African clades) co-circulating in this region, in which MDR phenotypes are common.13
Ampicillin, chloramphenicol and trimethoprim–sulfamethoxazole (co-trimoxazole) have traditionally served as first-line treatment for iNTS and typhoid fever in Africa.14 The widespread use of these antimicrobials likely led to the emergence and spread of MDR iNTS organisms. Several sub-Saharan African countries have reported MDR iNTS, including Malawi,15–17 Kenya,18–20 Ghana,21 Gambia,5 Democratic Republic of the Congo (DRC),22 Mozambique,23 24 Tanzania,25 Burkina Faso,7 Guinea-Bissau7 and Nigeria.26 Consequently, alternative treatments, such as ciprofloxacin, azithromycin and ceftriaxone, are used increasingly to manage bloodstream infections. However, these drugs may be unavailable or costly, and resistance is emerging to these antimicrobial agents.17 Concerningly, extensively drug-resistant (XDR) (MDR plus resistance to fluoroquinolones and third-generation cephalosporins) S. Typhimurium ST313 organisms have been reported in Kenya,14 27 Malawi28 and DRC.29 These new resistance phenotypes pose a significant challenge for the control of iNTS disease.14
Here, we subjected a contemporaneous collection of iNTS organisms from multiple sites in sub-Saharan Africa to whole genome sequencing to investigate the phylogenetic distribution of these organisms and their corresponding sequence types (STs) and antimicrobial resistance (AMR) determinants. We also estimated the incidence rates of MDR iNTS disease in the sampling locations and performed phylogenetic analyses of S. Typhimurium ST313 and S. Enteritidis ST11 in a global context.
Methods
Ethics approval and consent to participate
This research was conducted under the ethical principles of the Declaration of Helsinki. The IVI Institutional Review Board (IRB), the national ethical review committees in each participating country, and local research ethics committees approved the study protocol. All eligible patients meeting the study inclusion criteria were provided with a detailed explanation of the study purpose, and written informed consent was obtained prior to study enrolment. For children, written informed consent was obtained from parent or guardian.30
Study design and inclusion criteria
The majority of iNTS isolates (117/166) in this study originated from the Typhoid Fever Surveillance in Africa Program (TSAP),30 conducted in 13 sites in 10 countries between 2010 and 2014. Febrile patients from all age groups (except in Ghana, where only children aged <15 years were enrolled) with a tympanic or axillary temperature of ≥38.0℃ or ≥37.5℃, respectively, living in a defined study catchment area were eligible for recruitment. For inpatients, reported fever within 72 hours of admission was also necessary for inclusion. Written informed consent/assent was obtained. Clinical assessments of patients included history of illness, physical examination and clinical appraisal. Blood samples (5–10 mL for adults; 1–3 mL for children) were collected for microbiological testing and diagnosis. An additional 49 iNTS isolates were obtained from a febrile surveillance study conducted at the Presbyterian Hospital of Agogo in Ghana, between 2007 and 2009.31
Patient and public involvement statement
This TSAP study was performed under a single protocol with some site-specific and country-specific details; these details were developed with the study sites and local patient groups and communities attending the healthcare facilities. These data were essential for establishing the demographic framework of the sites and understanding how patients accessed healthcare and disease diagnosis. Therefore, these patient/community groups informed the study design to address issues regarding healthcare access and practices. Local communities and patients acted as communicators in the local communities to encourage people to attend healthcare facilities when symptomatic. Data from the studies have been provided to study sites to inform the local community and patients of the study findings.
Bacterial isolates and antimicrobial susceptibility testing
Blood specimens were inoculated into an aerobic blood culture bottle and incubated in systems with automated growth detection (BACTEC Peds Plus Medium/BACTECT Plus Aerobic-F, BACTEC, Becton-Dickinson, New Jersey; or BacT/ALERT PF Paediatric FAN/BacT/ALERT FA FAN Aerobic, bioMerieux, Marcy l’Etoile, France). Blood cultures with bacterial growth were sub-cultured on blood and chocolate agar (Oxoid, Basingstoke, UK), and biochemical tests were conducted (API 20E; bioMerieux) to identify suspected Salmonella isolates.30 Antimicrobial susceptibility testing was performed using agar diffusion tests according to the Clinical Laboratory and Standards Institute guidelines.30
Data sources and bacterial isolates
A total of 166 iNTS isolates were used for this investigation, which comprised 94 iNTS isolates from the TSAP study, an additional 23 iNTS isolates obtained from outside predefined TSAP study catchment areas and 49 iNTS isolates from other studies. In order to place these S. Typhimurium and S. Enteritidis isolates into a global phylogenetic context, the existing datasets were incorporated: 147 iNTS serotype Typhimurium ST313 isolates from seven countries (Malawi, Kenya, Mozambique, Uganda, DRC, Nigeria and Mali),11 Nigeria and DRC,12 Malawi,28 Kenya,14 Malawi32 and 594 iNTS serotype Enteritidis ST11 isolates (selected from Feasey et al13; online supplemental table 1).
Supplemental material
Whole genome sequencing
Genomic DNA was extracted from all Salmonella isolates using the Wizard Genomic DNA Extraction Kit (Promega, Wisconsin, USA). Two micrograms of genomic DNA from each organism was subjected to indexed-tagged pair-end sequencing on an Illumina Hiseq 2000 platform (Illumina, CA, USA) at the Wellcome Sanger Institute to generate 100 bp paired-end reads. Data quality control was performed using in-house pipelines. Raw sequence data are available in the European Nucleotide Archive (Project number: ERP009684, ERP010763, ERP013866) (online supplemental table 2).
Supplemental material
Single nucleotide polymorphism (SNP) calling and analyses
Raw Illumina reads were used to create multiple assemblies using Velvet V.1.233 with parameters optimised using VelvetOptimiser V.2.2.5,34 35 and automated annotation was performed using PROKKA V.1.5.36 Roary37 was used to define the pan genome of 166 iNTS isolates with blastp percentage identity of 99% and a core definition of 99%. In total, 3450 core genes were identified (genes that were present in ≥99% strains) and 86 765 SNP sites were extracted from the core gene alignment using SNP-sites V.2.1.3.35
For S. Typhimurium ST313, raw Illumina reads of 99 isolates from this study and 147 S. Typhimurium ST313 from previous studies11 12 29 33 38 were mapped to the reference sequence of S. Typhimurium strain SL1344 (accession: FQ312003.1), using SMALT V.0.7.4 (http://www.sanger.ac.uk/resources/software/smalt/). Candidate SNPs were called against the reference sequence using SAMtools38 and filtered with a minimum mapping quality of 30 and minimum consensus base agreement of 75%. The allele at each locus in each isolate was determined by reference to the consensus base in that genome using SAMtools mpileup and removing low confidence alleles with consensus base quality ≤20, read depth ≤5 or a heterozygous base call. The repeat finding program in NUCmer V.3.139 was used to identify exact repetitive regions of ≥20 bp in length in the reference genome and SNPs called in these regions were excluded. SNPs called in phage sequences and recombinant regions identified using Gubbins40 were further removed, resulting in a final set of 1960 chromosomal SNPs. The identification of SNPs for S. Enteritidis ST11 was performed following the same procedure as S. Typhimurium ST313. Briefly, the raw Illumina reads of 28 S. Enteritidis ST11 isolates from this study and 594 additional isolates from a global collection13 were mapped to the reference sequence of S. Enteritidis strain P125109 (accession: NC_011294.1), using SMALT followed by SNP calling and filtering as described previously, resulting in a final set of 25 121 SNPs.
Phylogenetic analyses
A maximum likelihood (ML) phylogenetic tree was constructed from the 86 765 SNP alignment of all 166 iNTS isolates using RAxML V.8.2.8 with a generalised time-reversible model and a Gamma distribution to model the site-specific rate variation (GTRGAMMA).41 Clade support for this tree was assessed through a bootstrap analysis with 100 pseudo-replicates. To investigate the molecular epidemiology of the S. Typhimurium ST313 and S. Enteritidis ST11 isolates sequenced here in a global context, a ML tree was inferred from an alignment of 1960 SNPs for 246 S. Typhimurium ST313 (99 from this study and 147 from previous studies11 12 29 33 38) and an alignment of 25 121 SNPs for 622 S. Enteritidis ST11 isolates (28 from this study and 594 from a global collection13), using RAxML with GTRGAMMA model. Support for these phylogenetic trees was assessed through a 100 bootstrap pseudo-analysis. Tree annotation was visualised using ITOL.42
Antimicrobial resistance gene and plasmid analyses
From raw Illumina reads, Short Read Sequence Typing-SRST243 was used to identify acquired AMR genes and their precise alleles using the ARG-Annot database,44 as well as plasmid replicons using the PlasmidFinder database.45 Multilocus sequence typing (MLST) of all iNTS isolates was also determined using SRST2 together with the MLST database for Salmonella enterica downloaded from pubMLST (https://pubmlst.org/organisms/salmonella-spp).46 Salmonella serotypes were identified using conventional serology as well as MLST-based approach47 and SeqSero (genome-based approach)48; the final interpretation followed a consensus of MLST and SeqSero. Bandage49 was used to investigate the assembled contigs carrying the AMR cassettes. Mutations in fluoroquinolone resistance genes (gyrA, gyrB, parC, parE) were identified using SeaView.50
Incidence analyses of MDR iNTS disease
Incidence rates of MDR iNTS were estimated for study sites in Burkina Faso, Ghana, Guinea-Bissau, Kenya and Senegal. Statistical methodology used previously to calculate the incidence of S. Typhi and iNTS disease in the TSAP study was used to calculate MDR iNTS incidence.7 30 51 Demographic data from Health and Demographic Surveillance System (HDSS) sites were used to estimate population denominators where available. In non-HDSS sites, health-seeking behaviour as reported by randomly administered healthcare utilisation surveys were used to estimate population denominators for each study site. Crude incidence rates were adjusted to account for the proportion of surveyed individuals who reported seeking care for a febrile episode at a study facility to yield adjusted incidence rates. Adjusted incidence rates of MDR iNTS cases per 100 000 per person-years-of-observation (PYO) were estimated with 95% CIs using these adjustment factors and crude MDR iNTS case numbers. The previously established multi-country database (Microsoft Visual FoxPro 7.0, Redmond, Washington) for TSAP was used for the countries with MDR iNTS isolates.
Results
Geographical distribution of iNTS serotypes and sequence types
The majority (66%; 110/166) of iNTS organisms in the sampled sub-Saharan African countries were S. Typhimurium, of which 90% (99/110) were ST313 and 10% (11/110) were ST19. S. Enteritidis accounted for 18% (30/166) of the isolates, composed of ST11 (93.3%; 28/30), ST183 (3.3%; 1/30) and ST2107 (3.3%; 1/30). S. Dublin (ST10) comprised a further 11% (18/166) of isolates. Several other serotype STs, including S. Choleraesuis ST145 (3/166), S. Muenster ST321 (1/166), S. Poona ST308 (1/166), S. Stanleyville ST339 (1/166) and S. Virchow ST359 (1/166), were also identified. S. Typhimurium ST313 was mostly limited to West Africa (table 1), whereas S. Enteritidis ST11 appeared to be pervasive in both West and Southern Africa. S. Dublin ST10 was identified in West Africa and S. Typhimurium ST19 was distributed across the continent.
Distribution of iNTS serovars and genotypes circulating in the sampled countries in sub-Saharan Africa
Overall, 61% (102/166) of the iNTS organisms described here were MDR. These were isolated in Burkina Faso (83%; 10/12), Ghana (66%, 88/133), Guinea-Bissau (22%; 2/9), Kenya (100%; 1/1) and Senegal (50%; 1/2). S. Typhimurium exhibited the highest prevalence of MDR (85%; 94/110); 95% (94/99) of the ST313 isolates were MDR. In total, 23% (7/30) of S. Enteritidis and 6% (1/18) S. Dublin organisms were MDR; none of the S. Typhimurium ST19 were MDR (figure 1, table 2).

Geographical distribution of iNTS genotypes and serotypes in the sampled countries in sub-Saharan Africa. Different colours in the pie charts correspond to different sequence types and serovars of iNTS isolates in our study sites. The size of the pie charts corresponds to the numbers of isolates in each country. Countries coloured in grey highlight the study sites, while red and white circles indicate countries with and without MDR iNTS isolates, respectively.
Distribution of MDR iNTS and gyrA mutation (fluoroquinolone resistance) in the sampled countries in sub-Saharan Africa
Phylogenetics and AMR of iNTS
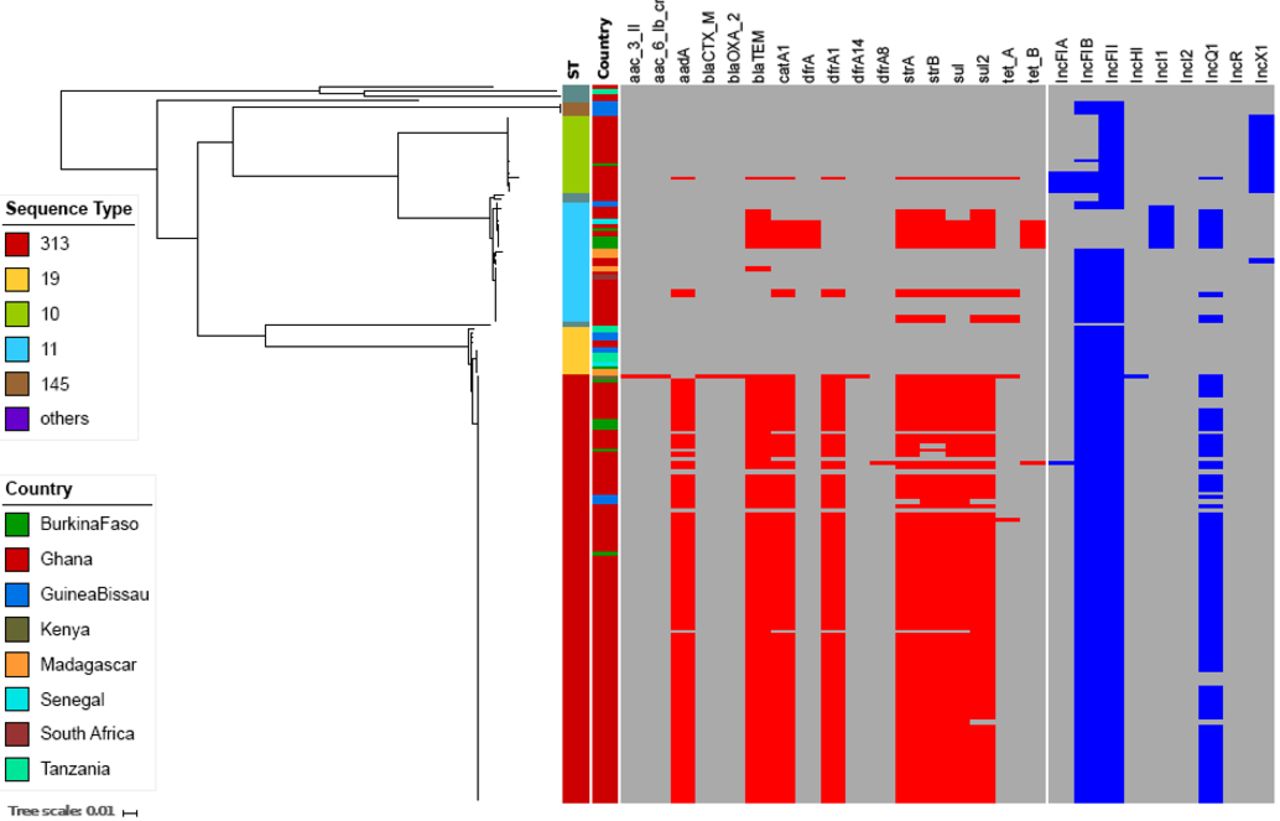
A phylogenetic reconstruction of all iNTS isolates showed that the three major serovar STs—S. Typhimurium (ST313), S. Enteritidis (ST11) and S. Dublin (ST10)—formed independent clusters with dissimilar AMR gene profiles (figure 2). Almost all of the MDR S. Typhimurium ST313 (95%; 94/99) carried the Tn21 transposon-associated MDR-loci (sulII-strAB-dfrA1-aadA1-sulI-cat-blaTEM) on an IncF virulence-resistance plasmid (pSLT-BT).11 A single MDR S. Typhimurium ST313 from Kenya additionally carried two copies of blaCTX-M-15, conferring resistance to third-generation cephalosporins. One copy of blaCTX-M-15 was located on the 300 kb IncHI2 plasmid, pKST313 (accession number: LN794248), and the other on the chromosome disrupting the ompD locus. Reduced susceptibility to fluoroquinolones in S. Typhimurium ST313 was uncommon, with 2% (2/99) of Ghanaian ST313 isolates possessing a single mutation (S87Y) in gyrA (table 2).

Antimicrobial resistance genes and plasmids associated with iNTS isolates circulating in sampled sub-Saharan African countries. Midpoint-rooted maximum likelihood phylogenetic tree based on the core genes of iNTS isolates sequenced in this study and their corresponding metadata. The first column shows the sequence types in different colours. The second column corresponds to the countries where our iNTS isolates were detected. The remaining columns exhibit a heatmap of detected AMR genes and plasmid replicons. The tree scale bar indicates the number of substitutions per variable site.
The majority of S. Enteritidis ST11 (19/28; 68%) harboured the typical IncF virulence plasmid (60 kb), which was comparable with pSENV (accession number NC_019120.1, coverage 100%, identity 99%). The remaining S. Enteritidis ST11 (9/28; 32%) harboured a novel IncI1 virulence-resistance plasmid (pSEP, accession number: ERP121368) of approximately 68 kb (figure 3), of which 7/28 (25%) isolates (4 from Burkina Faso, 2 from Ghana, 1 from Senegal) carried the MDR-encoding Tn21-like transposon (sulII-strAB-tetB-dfrA1-sulI-cat-blaTEM), and 2/28 (7.1%) isolates carried a different AMR cassette (TnpA-sulII-strAB-blaTEM-Tn3). The novel IncI plasmid exhibited 60% homology to pSENV but did not harbour the IncF replicon, the pefBACD fimbriae-encoding operon, or the virulence-associated genes srgA and rck (figure 3). In addition, two non-MDR Ghanaian S. Enteritidis ST11 possessed an AMR cassette (sul2-strAB-tetA) carried on a small (11 kb) non-conjugative IncQ plasmid conferring resistance against sulfonamides, streptomycin and tetracyclines. This IncQ plasmid exhibited a similar genetic structure to pSTU288-2 from S. Typhimurium (accession number CP004059.1, coverage 98%, identity 99%). Two further non-MDR Ghanaian isolates carried a Tn21-mediated AMR cassette (sulII-strAB-dfrA1-aadA1-sulI-cat) on the virulence plasmid, and a single non-MDR isolate from Madagascar carried a blaTEM Tn3 integrated into the virulence plasmid. Reduced susceptibility to fluoroquinolones was predicted by the sequences in 39% (11/28) of the S. Enteritidis-ST11, all of which originated from Ghana and displayed a variety of gyrA mutations (D87G: 6 isolates, D87N: 3 isolates, D87Y: 2 isolates) (table 2).

Novel IncI1 virulence-resistance plasmid (pSEP) in an S. Enteritidis ST11 isolate. Plasmid comparison analyses between the novel virulence-resistance IncI1 plasmid pSEP (bottom) and the reference virulence IncF plasmid pSENV (top). The grey blocks show the BLASTn comparison between the two plasmids using the bl2seq feature from the web-based BLAST. Some annotations are added for both plasmids. Red-coloured arrows are genes associated with virulence and AMR. Blue-coloured arrows are genes associated with conjugation. Grey-coloured arrows correspond to plasmid replication and stability. Green-coloured genes are associated with transposon elements.
Phylogenetics of S. Typhimurium ST313 and S. Enteritidis ST11 isolates in global context
To investigate the population structures of S. Typhimurium ST313 and S. Enteritidis ST11 in a broader context, we constructed global phylogenies. All S. Typhimurium ST313 isolates from our study fell into lineage II (figure 4A). The single Kenyan ST313 isolate carrying two copies of blaCTX-M-15 was part of the previously described clonal expansion of MDR ceftriaxone-resistant ST313 sub-lineage.14 Notably, the Ghanaian ST313 isolates did not form a single cluster, but were associated with multiple clusters from Mali, Burkina Faso and Nigeria, indicating multiple introduction events (figure 4A).
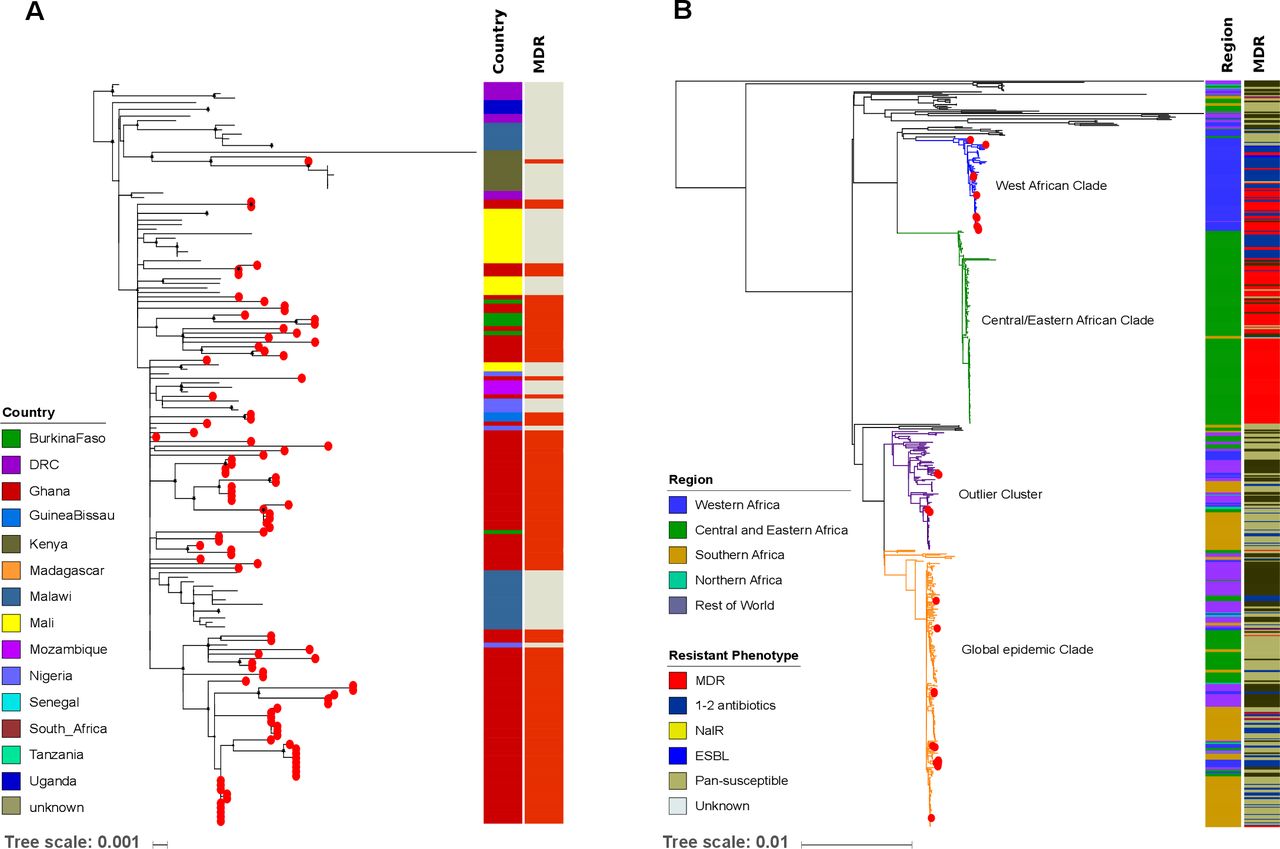
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Phylogenetics of S. Typhimurium ST313 lineage II and S. Enteritidis ST11 in sub-Saharan Africa. (A) Maximum likelihood phylogenetic tree of S. Typhimurium-ST313 lineage II isolates from this study in the context of the sub-Saharan African continent; lineage I was pruned to enhance the visualisation of the tree. Red circles at the terminal leaves correspond to our study isolates. The first column shows different colour-coded countries from where all analysed isolates originate. The second column shows MDR and non-MDR isolates in red and grey, respectively. (B) Maximum likelihood phylogenetic tree of our S. Enteritidis-ST11 isolates in the global context of S. Enteritidis. Red circles at the terminal leaves correspond to our study isolates. The first and second columns show regions and resistant phenotypes in different colours. The tree scale bar indicates the number of substitutions per variable site.
A detailed phylogenetic investigation of S. Enteritidis ST11 demonstrated that 11/28 (40%) (Ghana: 5, Burkina Faso: 4, Senegal: 1, Guinea Bissau: 1) isolates fell into the West African clade.13 The ST11 organisms within this West African clade displayed either MDR (seven isolates) or other non-MDR AMR phenotypes (two isolates). We found evidence that some Ghanaian isolates within this clade clustered alongside organisms from Burkina Faso and Mali, again suggesting international transmission. In addition, 13/28 (46%) of the S. Enteritidis ST11 (Ghana: 11, Madagascar: 1, South Africa: 1) belonged to the Global epidemic clade.13 These isolates had phylogenetic links with their country-specific clusters, with the exception of two Ghanaian isolates that clustered with organisms from neighbouring Cameroon and Senegal. Lastly, 4/28 (14%) (2 from Ghana, 2 from Madagascar) of the ST11 isolates grouped within the outlier cluster (figure 4B).
Incidence of MDR iNTS disease in sub-Saharan Africa
We calculated the age-stratified incidence rates of MDR iNTS in previously described study catchment areas in Burkina Faso, Ghana, Guinea-Bissau and Kenya (table 3). The adjusted incidence of MDR iNTS disease exceeded 100/100 000 PYO in children <15 years of age in all West African countries: Burkina Faso (Nioko II, 274/100 000 PYO, 95% CI 185 to 406; Polesgo, 255/100 000 PYO, 95% CI 138 to 470), Ghana (Asante Akim North: Ghana-AAN, 414/100 000, 95% CI 333 to 515) and Guinea-Bissau (Bandim, 105/100 000, 95% CI 69 to 161). Among children <15 years, younger children (<2–4 years) exhibited the highest MDR iNTS incidence rates in both sites in Burkina Faso: 753/100 000 PYO (95% CI 460 to 1233) in Nioko II and 630/100 000 PYO (95% CI 288 to 1380) in Polesgo. In both settings in Burkina Faso, the incidence of MDR iNTS disease in the infant age group was slightly lower than in the group aged 2–4 years, but children <5 years old exhibited a high burden of MDR iNTS disease. In Ghana-AAN, infants aged <2 years had the highest incidence of MDR iNTS disease (1435/100 000; 95% CI 1110 to 1854) followed by children aged between 2 and 5 years (747/100 000; 95% CI 491 to 1135). Similarly, in Guinea-Bissau, infants <2 years old exhibited the highest incidence of MDR iNTS disease (291/100 000; 95% CI 176 to 482). The incidence rate of MDR iNTS in older age groups (≥15 years) was relatively low, ranging between 0 (Guinea-Bissau) to 11 (Kenya) and 54 (Burkina Faso) per 100 000 PYO.
Incidence estimates of MDR iNTS disease in sub-Saharan Africa
All iNTS organisms analysed
Discussion
Our study shows that MDR iNTS is highly prevalent in several sub-Saharan African countries. Specifically, we found that MDR S. Typhimurium ST313 is the most common cause of iNTS disease, but that other iNTS serovars, principally S. Enteritidis ST11 and S. Dublin ST10 in West Africa, also constitute a major proportion of the disease burden.52 53 Our phylogenetic analyses provide further evidence for the regional transmission of two MDR serovars/STs (S. Typhimurium ST313 and S. Enteritidis ST11) between Ghana and neighbouring countries Burkina Faso, Nigeria, Mali and Senegal. These transmission events highlight the need for intensified AMR surveillance, the coordination of AMR reporting, and sustained public health control measures between these and other African countries.
We calculated a particularly high incidence of MDR iNTS disease in the West African countries of Burkina Faso, Ghana (<5 years) and Guinea-Bissau (<2 years). The incidence rates of MDR iNTS disease presented here generally correspond with the incidence of iNTS disease in other African countries.7 Effective antimicrobial therapy is an essential component of iNTS management; however, the effectiveness of first-line treatments has been diminished due to the emergence and spread of MDR and XDR NTS strains.13 29 54 Our data also depict the emergence of reduced fluoroquinolones susceptibility in both MDR ST313 and ST11 in Ghana, as well as the circulation of a ceftriaxone-resistant ST313 sub-lineage in Kenya. Invasive Salmonella with reduced susceptibility to ciprofloxacin have been reported in Burkina Faso,7 Ghana,7 21 Nigeria,55 Senegal,56 Mozambique,23 the Congo,29 57 Kenya58 and South Africa.59 This increasing trend in resistance against clinically important classes of antimicrobials in differing iNTS serovars across Africa is of major concern.14 The growing use of ceftriaxone and ciprofloxacin for the treatment of febrile illnesses in Africa may lead to an increase in of MDR and XDR pathogens in this continent, which has already been observed across Asia in the last two decades.
Several limitations should be considered in interpreting and generalising these data beyond our study sites. While we are able to illustrate the magnitude of the problem of MDR iNTS in West Africa, there are relatively few genomic data points available from countries in East and Southern Africa. Previous studies showed high prevalence of MDR iNTS disease in Kenya27 58 and a recent meta-analysis suggested that MDR iNTS has emerged across four regions of sub-Saharan Africa.60 The generation and analysis of additional epidemiological and genomic iNTS data in Eastern/Southern Africa would help facilitate comparison of incidence rates and AMR profiles of iNTS-associated organisms between African regions. Further, the estimated incidence rates of MDR iNTS disease in our study should be interpreted with caution as the number of cases in some countries were relatively small. In parallel, the original study design may also have led to some underestimation of iNTS burden, as afebrile patients with other clinical symptoms associated with iNTS disease were not enrolled. As a result, the true incidence of MDR iNTS disease in some settings need to be further monitored with more systematic disease surveillance.
Despite the identified limitations, our study provides enhanced insights into the population structure and transmission dynamics of major MDR iNTS serovars in sub-Saharan Africa and identified countries with a high burden of MDR iNTS. There is an urgent need to expand clinical and genomic surveillance for pathogens causing bloodstream infections across continental Africa to improve our understanding of disease incidence and to monitor AMR trends. Such data can better inform antimicrobial stewardship to extend the life of existing antimicrobial therapies and prioritisation of preventative interventions including vaccines. The development and deployment of a safe, low-cost, highly efficacious multivalent vaccine should be prioritised for the management and prevention of iNTS disease in Africa, particularly in countries with high prevalence of MDR iNTS infections, as well as HIV, malaria and malnutrition.61 62 Meanwhile, further investigations of household transmission dynamics and human and non-human reservoirs of infection are warranted to inform better iNTS control measures and, ultimately, optimal programmatic use of future vaccines.
Data availability statement
Data are available in a public, open access repository. Raw sequence data are available in the European Nucleotide Archive (Project number: ERP009684, ERP010763, ERP013866).
Ethics statements
Acknowledgments
We would like to acknowledge and thank all staff and partners involved in obtaining and processing the data and samples including healthcare facility and laboratory staff at the TSAP network countries. We also thank the WTSI Pathogen Informatics team for help with whole genome sequencing.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Handling editor Seye Abimbola
Twitter @DrSeEunPark, @DuyPham_Oucru, @meganecarey
SEP and DTP contributed equally.
Contributors Conceiving and designed experiments: SEP, GD, SB, FM. Performed experiments: SEP, DTP. Analysed the data, contributed reagents/materials/analysing tools: SEP, DTP, GDP, UP, LMCE, VvK, JI, ODM, HSG, JAP, RFB, YAS, EOD, RR, ABS, AA, NG, AS, KHK, JM, PA, HMB, JTH, JMM, LC, BO, BF, NS, TJLR, TMR, LPK, ES, MT, BY, MAET, RK, DMD, AJ, AT, AN, MBA, SVL, JFD, JKP, FK, MEC, SVP, MA, JDC, GD, SB, FM. Funding acquisition: GD, FM. Writing original draft: SEP. Writing review and editing: SEP, DTP, SB, FM.
Funding This work was supported by the Bill & Melinda Gates Foundation (grant: OPPGH5231). The findings and conclusions are our own and do not necessarily reflect positions of the Bill & Melinda Gates Foundation or the US Centers for Disease Control and Prevention. The International Vaccine Institute acknowledges its donors, including the Government of Republic of Korea and the Swedish International Development Cooperation Agency (SIDA). Research infrastructure at the Moshi site was supported by the US National Institutes of Health [grant numbers R01TW009237; U01 AI062563; R24 TW007988; D43 PA-03-018; U01 AI069484; U01 AI067854; P30 AI064518), and by the UK Biotechnology and Biological Sciences Research Council [grant number BB/J010367]. Stephen Baker is supported by a Wellcome senior research fellowship (215515/Z/19/Z). DTP is funded as a leadership fellow through the Oak Foundation [grant number OCAY-15-547].
Disclaimer The funders had no role in the design and conduct of the study; collection, management, analysis, and interpretation of the data; preparation, review, or approval of the manuscript; and decision to submit the manuscript for publication.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.