Article Text

Abstract
Implementing an Ebola vaccine trial in a remote area in the Democratic Republic of the Congo (DRC), and being confronted with a dysfunctional health care system and acute unmet health needs of participants, ethical considerations were made regarding the ancillary care obligations of the sponsor and researchers. Spurred by the occurrence of non-related (serious) adverse events (NR-SAEs), the Universities of Antwerp and Kinshasa jointly developed an algorithm, accompanied by an algorithm policy. The algorithm consists of a set of consecutive questions with binary response options, leading to structured, non-arbitrary and consistent support and management for each NR-SAE. It is the result of dialogue and collaboration between the sponsor (University of Antwerp) and the principal investigator (University of Kinshasa), consultation of literature, and input of research ethics and social sciences experts. The characteristics of the project and its budgetary framework were taken into account, as well as the local socioeconomic and healthcare situation. The algorithm and related policy have been approved by the relevant ethics committee (EC), so field implementation will begin when the study activities resume in November 2021. Lessons learnt will be shared with the relevant stakeholders within and outside DRC.
If NR-SAEs are not covered by a functioning social welfare system, sponsors and researchers should develop a feasible, standardised and transparent approach to the provision of ancillary care. National legislation and contextualised requirements are therefore needed, particularly in low/middle-income countries, to guide researchers and sponsors in this process. Protocols, particularly of clinical trials conducted in areas with ‘access to care’ constraints, should include adequate ancillary care arrangements. Furthermore, it is essential that local ECs systematically require ancillary care provisions to enhance the well-being and protection of the rights of research participants. This project was funded by the European Union’s Horizon 2020 research and innovation programme, European Federation of Pharmaceutical Industries and Associations, and the Coalition for Epidemic Preparedness Innovations.
- clinical trial
- health insurance
- health policy
- public health
Data availability statement
All data relevant to the study are included in the article.
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Summary box
Ancillary care guidelines are often theoretical rather than descriptive, and until today remain non-binding.
Contextualised legislation and clear national requirements are needed, especially in countries without a functioning social welfare system, to ensure that ancillary care is integrated in clinical trials involving socioeconomically vulnerable individuals and communities.
Ethics committees should explicitly require adequate and well-implemented ancillary care provisions.
An algorithm and algorithm policy is presented that can provide a practical way to non-arbitrarily address ancillary care needs in a vaccine clinical trial, and that can be adapted to the specificities of other clinical trials in settings with access to care constraints.
Findings, adaptations and lessons learnt coming from the operationalisation of this algorithm and algorithm policy will be documented and shared with relevant stakeholders within and beyond the Democratic Republic of Congo.
Introduction
In the years that followed its independence in 1960, the Democratic Republic of the Congo (DRC) became a pioneer in developing the health district model in SubSaharan Africa. Unfortunately, the quality of health services rapidly declined since the mid-70s, and the health system performance was severely affected by the collapse of the state and economy that followed at the beginning of this century. The consequent lack of government funding of the health sector led to poor access to healthcare, a deterioration of the quality of services and acute unmet health needs.1 As a health insurance system is not widespread, patients must pay for services out-of-pocket at the point of care.2 3 Such direct payments are considered the most regressive and inequitable mode of financing healthcare, claiming a higher proportion of income among poor households.4 5
Additionally, DRC is the most Ebola-affected country. It declared the end of its 12th Ebola outbreak in May 2021.6 Various Ebola trials have been conducted in DRC as clinical research is an essential component of outbreak response and preparedness. Currently a Phase 2 clinical trial is being conducted in Boende, a town situated in a remote, potentially Ebola endemic region in the northwest of DRC.7 Boende is the capital of the Tshuapa Province and experienced an outbreak in 2014.8 Under the sponsorship of the University of Antwerp (UAntwerp) and with the principal investigator (PI) based at the University of Kinshasa (UNIKIN), the trial aims to improve preparedness for future outbreaks by vaccinating a cohort of healthcare providers. As described in the Clinicaltrials.gov registry (NCT04186000), recruitment started in December 2019. A first vaccine was administered, followed by a second dose 57 days later. Depending on the randomisation group, a booster vaccination is administered 1 or 2 years post first dose. The trial is scheduled to end in October 2022.
As vaccines have the potential of causing adverse events (AEs), the trial was set up to manage all AEs equally, regardless of relatedness to the experimental vaccine. However, the practical implementation of this approach in the study context was not straightforward for serious AEs (SAEs). Identifying and reporting SAEs is a key regulatory requirement.9 Furthermore, providing adequate care for SAEs is an essential ethics requirement, framed in the broader obligation to make adequate provisions for participants’ health needs—based on the extent to which they need assistance, and effective care is locally available and affordable.10 Nonetheless, there is an ongoing debate on sponsors’ and researchers’ obligations when conducting ‘good clinical practice (GCP) compliant health research’ in low/middle-income countries (LMICs), or in other low-resource settings where participants have limited access to care through the local health system.11 In particular, this debate is centred around the concept of ‘ancillary care’ (AC), which is defined by the Council for International Organizations of Medical Sciences (CIOMS) as the care provided during research, when participants develop conditions other than those related to the study.10 Several examples of the need to provide AC12 or of good practices in doing so,11 clearly indicate that unmet health needs are a recurrent issue.
Though the awareness of AC needs has grown, discussion continues on how normative models and ethical guidance on AC can be translated into feasible practices.13 To the best of our knowledge, there are no internationally binding requirements of AC in clinical trials. However, national legislations might require compensatory mechanisms for injuries unrelated to research. For example, since 2013 India applies a ‘no fault’ compensation principle whereby all injuries, related and unrelated to a trial, are to be compensated entirely by the sponsor.14 This legal regimen has far-reaching implications for both internationally and locally led research in India. In the DRC, conversely, there are no specific requirements or legislation requiring sponsors to provide AC.
Our research group was involved in several LMIC-based studies, among them major non-commercial phase 3 trials, whereby free care was offered on site for all SAEs, irrespective of relatedness to the investigational product (IP).15 16 The rationale was both methodological—that is, excluding participants with NR-SAEs may cause an incorrect or varying causality assessment—and ethical—that is, participants in the same study receive a similar approach to care, independent of the causality of the SAE. Furthermore, the causality assessment may not be definite and can be reviewed when data accumulate.
However, an on-site causality assessment is not always feasible. For instance, in the current Ebola trial, participants are followed up for several years, often reside at long distances from the study site, and have poor transport availability. Therefore, a consistent, non-arbitrary approach was needed, to fairly and equally support the participants for their SAE-related expenses, whether made on site or at another (remote) health centre. To address this need, we developed a decision-making algorithm, and framed it into a comprehensive algorithm policy.
Development process
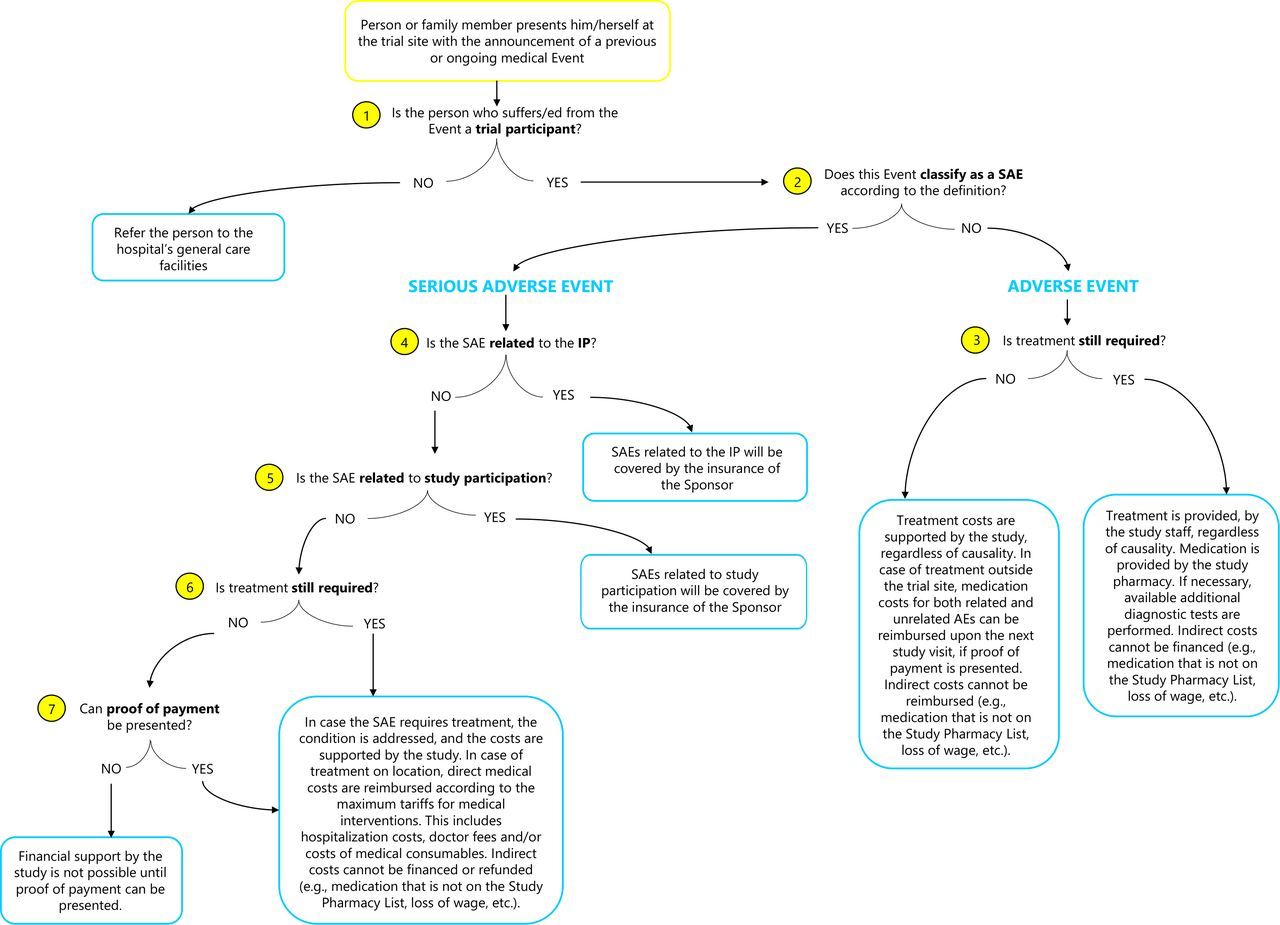
Definitions of an AE and SAE used in the algorithm (figure 1) were obtained from the International Conference for Harmonization (ICH) of Technical Requirements for Pharmaceuticals for Human Use17: (1) an AE is any untoward medical occurrence in a participant who received an IP which does not necessarily have a causal relationship with the IP; (2) an SAE ‘is any untoward medical occurrence that at any dose results in death; is life-threatening; requires inpatient hospitalisation or results in prolongation of existing hospitalisation; results in persistent or significant disability/incapacity; is a congenital anomaly/birth defect; is a medically important event or reaction’.

Visual representation of the algorithm to approach non-related (serious) adverse events in an Ebola vaccine trial in a remote setting, Boende, DRC. This figure outlines the chronological set of questions (‘inclusion or exclusion criteria’) that lead from a given initial state (‘the occurrence of a medical event’) to an intended goal (‘an approach and outcome for each individual case’). The event that presents itself to the study team is visualised by a yellow box. The questions that guide the decision-making process are indicated by a yellow circle containing a number. The lines after each question represent the binary response, leading to either an outcome for that individual case, presented in a blue box, or a follow-up question to continue the sequence. DRC, Democratic Republic of the Congo; IP, investigational product; SAE, serious adverse event.
The development of the algorithm and algorithm policy (online supplemental material) was prompted by the manifestation of six NR-SAEs during the recruitment phase of the 699 trial participants (December 2019 to February 2020) (table 1). These events happened nearby or at distance from the study site. As such, some participants first sought treatment locally and later reported the event to the study team. Although the participants had no other choice, this sequence made it difficult for the study team to assess their medical needs, the causality of the event and the financial support they could offer. The extent to which the study would provide support in any (NR-)SAE was not easily determined. A consistent and non-subjective guidance tool seemed essential.
Supplemental material
Six initial non-related serious adverse event (NR-SAE) cases
The development process (figure 2) of the algorithm and algorithm policy was thus steered by the experiences with these first NR-SAEs. As the initial events were managed ad hoc by the PI, a systematic approach became imperative. Consequently, a PubMed search was performed between March and July 2020 that initially focused on finding guidance and/or a decision-making algorithm applied in other clinical studies in LMICs to approach NR-SAEs. Words in the search string included but were not limited to: ‘algorithm’, ‘adverse event’, ‘serious adverse event’, ‘non-related serious adverse event’, ‘NR-SAE’, ‘SAE unrelatedness’, ‘injured research subjects’, ‘compensation unrelated injuries’ and so on. During this search, no algorithm or hands-on approach from other studies was found or published. Also, as no specific AC requirements or expectations were issued by the Ministry of Health in the DRC, the sponsor and PI consulted literature to guide and develop such an algorithm. A purposive search of documents such as Policy documents, ethics guidelines, WHO guidelines, regulatory guidelines on AC practices and responsibilities in public health (from LMICs) and so on was performed. Literature discovered during the PubMed and purposive search that was useful to develop the algorithm was combined. On these relevant sources, authors were also directed by references of references (snowballing method) whereby original research was consulted to further explore guidance. Finally, the lead author also followed the Ethics of Ancillary Care in Research E-course, provided by The Global Health Network (2020),18 which also presented useful sources. In the end, normative models and general ethical guidance on AC steered the development of the algorithm and concomitant algorithm policy, keeping in mind the principle of beneficence,10 and the obligation of researchers to maximise benefit and minimise harm for participants.
{kind=link}
{kind=link}
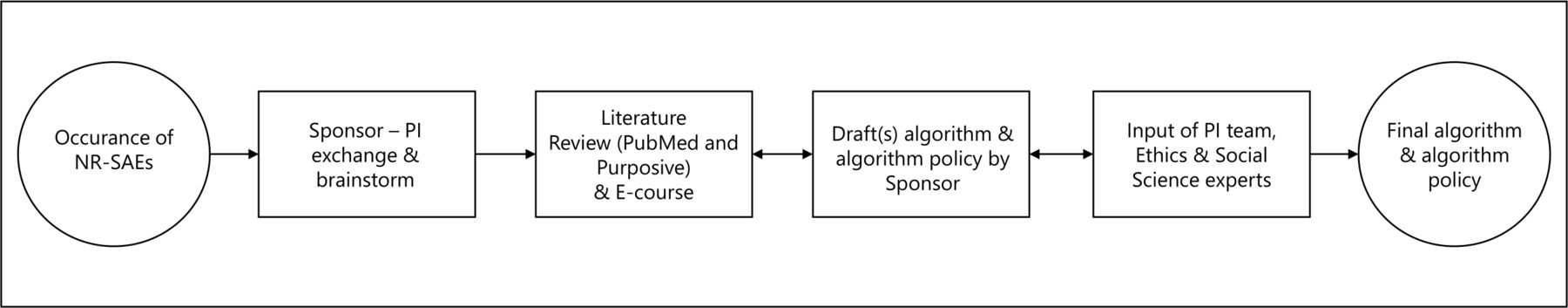
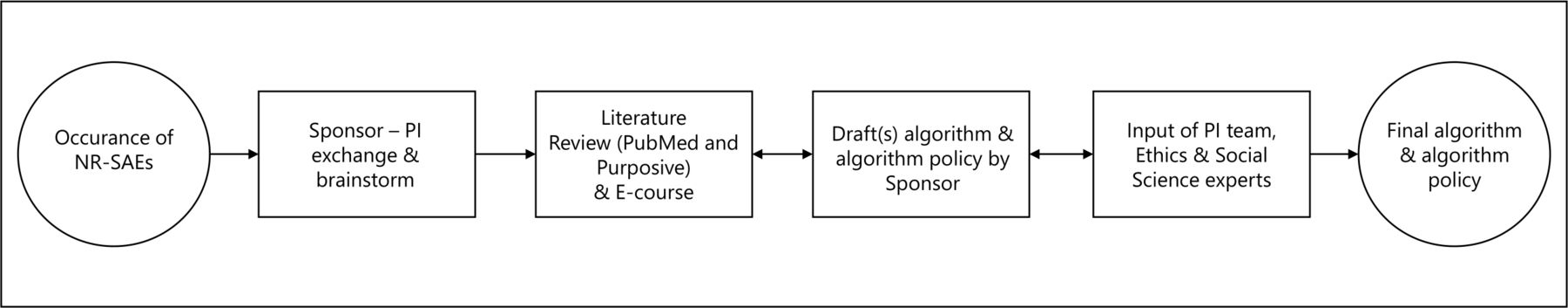
Development process of the non-related (serious) adverse events algorithm and algorithm policy. This figure outlines the development process of an algorithm and algorithm policy for an Ebola vaccine trial in DRC. The process started with the occurrence of NR-SAEs, also represented in the first circle, which led to several steps, visualised by a square, initiated by the sponsor. A single arrow refers to a consequent action. A double arrow indicates a reciprocal relationship between steps. The outcome, a finalised algorithm and algorithm policy, is represented by the last circle. DRC, Democratic Republic of the Congo; NR-SAE, non-related serious adverse event; PI, principal investigator.
The algorithm was gradually built up via a set of questions (‘inclusion or exclusion criteria’) that led from an initial fact (‘the occurrence of a medical event’) to an intended goal (‘an approach to each individual case’) via binary response options. Several drafts were shared and discussed within the research team at both universities. The proposed criteria were frequently questioned, debated, changed and/or restructured with respect to their relevance, precedence and priority. Being DRC nationals, the PI team had excellent knowledge and understanding of the local circumstances in Boende, that do not differ much from other remote locations in the country. Therefore they were considered to represent the viewpoints and input of local representatives. The algorithm was thus developed in close collaboration between the sponsor and PI team by having frequent meetings until agreement was reached on the algorithm’s approach and the translation of the algorithm policy into practice.
Once a consensus on the main framework of the algorithm policy was reached, social sciences and research ethics experts were consulted for their expertise, whereby one member was affiliated with the PI’s institution (Member of the Ethics committee of the School of Public Health at UNIKIN) and two members had links with the sponsor (a One Health anthropologist at UAntwerp and the chair of the Institutional Review Board of the Antwerp Institute of Tropical Medicine). Their input was critical in addressing the remaining ethical challenges of approaching AC needs and their practical elaboration within this study. Finally, the algorithm policy was submitted to the National Committee of Health Ethics in DRC for review. Approval was granted on 1 March 2021 (n°231/CNES/BN/PMMF/2021).
The algorithm and algorithm policy
Figure 1 represents the algorithm to approach SAEs in the Ebola vaccine trial in Boende, DRC. Each event that presents itself is assessed case-by-case in terms of the required intervention and support. Below the modalities and operationalisation are addressed under each question.
Identifying beneficiaries
People who are not trial participants are referred to the hospital’s general care facilities. Only trial participants are beneficiaries. In this trial, identification is confirmed via a participant card and iris scanning. In the event, participants are asked to discontinue the study intervention, for example, due to a medical condition, they remain in the trial for safety follow-up. Therefore, they continue to benefit from the algorithm policy. Coverage stops when participants are lost to follow-up (figure 1, question 1).
Type of medical event
A distinction is made between AEs and SAEs. According to the protocol, hospitalisations planned prior to giving consent to participate in the trial, or existing conditions which have not worsened since enrollment, are not considered SAEs. Events resulting in the prolongation of (planned) hospitalisations are new SAEs (figure 1, question 2).
Adverse events
As vaccines might cause AEs, it was explained to participants during the informed consent process (prior to enrollment in the trial) which solicited AEs could occur, and on how to recognise them. Participants were also advised to report any AE to the site, whether mentioned during the informed consent or not. There is no distinct treatment approach between related or unrelated AEs. If the AE is ongoing, the participant’s condition is treated and if necessary available additional diagnostic tests are performed by the study staff. Medication is provided from the study pharmacy. The latter was based on the Model List of Essential Medicines of the WHO19 that aims to cover the minimum medicine needs for adequate healthcare provision and serves as a reference tool for public healthcare. Deviations therefrom in the study pharmacy were due to practical or context-specific considerations (eg, stock-outs, quantity adaptation to the size of the study, risk mitigation for expiry dates and so on) (figure 1, question 3).
If participants had been treated outside the trial site, medication costs for both related and unrelated AEs could be reimbursed on the next study visit, provided that proof of payment is presented, and the medications are on the Study Pharmacy List. It is considered that other medications do not address essential health needs and thus fall outside of the study’s support. Indirect costs caused by AEs are not supported by the study (eg, loss of wage).
Causality of the SAE
The causality to the IP is preliminarily assessed by the site PI or a delegated site medical doctor. This is a binary assessment. First, in line with the WHO handbook for GCP, all trial-related injuries will be covered by the insurance of the sponsor.20 This refers to the fourth question in the algorithm. Second, ‘trial-related’ can be interpreted as the result of study participation in a broader sense, as indicated by question five in the algorithm. For instance, a participant may get injured on the way to or from the trial site, or while on site. The modalities of what ‘relatedness to study participation’ entails, is a decision the sponsor and PI should make upfront, based on the local circumstances. In the present trial, this is defined as ‘all actions and displacements directly related to trial participation or during trial activities on site’. In case of unrelatedness, there is a reasonable likelihood that study participation or the administration of the IP did not cause the SAE. This is where the algorithm policy under discussion steps in (figure 1, Questions 4,5).
Status of the NR-SAE
When the NR-SAE is ongoing, the participant’s condition is treated. However, in this context with its remote setting, long distances and poor availability of transport, it is very plausible that a participant seeks urgent treatment outside the trial facilities, as was the case with some of the initially reported NR-SAEs (table 1). Relatedly, a participant could have been hospitalised elsewhere, was unable to inform the site at the moment the SAE occurred, or has deceased. Either the participant, or in the latter case a relative, reaches out to the trial staff with the announcement of an SAE treated in a different location. When treatment is no longer required (eg, was done elsewhere) or can no longer be given (eg, the participant has deceased), the policy can retroactively apply. An assessment of the reported SAE based on the input from the treating medical doctor (on the treatment, medications given and so on) should be obtained and assessed by the PI (figure 1, Questions 6,7).
Irrespective of the status of the NR-SAE, direct medical costs considered standard of care will be supported. This involves hospitalisation fees, medical interventions, doctor fees and/or costs of medical consumables. Transportation costs are also covered when related to the announcement or treatment of the NR-SAE. Additional transport arrangements can be made for a participant to continue with the trial activities (eg, participant with a leg fracture). Possible supplementary costs (eg, medication outside of the study pharmacy list) are not supported. Indirect costs resulting from possible long-term consequences of NR-SAEs cannot be covered as these fall outside of a research responsibility (eg, loss of wage, burial costs and so on).
Unrelated to and well before the start of the Ebola vaccine trial, each local healthcare provider in the Boende health district set a list with maximum tariffs for medical interventions. The list of the General Reference Hospital of Boende served as a reference for the reimbursement of costs when an SAE is treated outside of the trial facilities, provided that the SAE is confirmed and the medical interventions are assessed to be related. Additionally, proof of payment is a requirement for reimbursement, as indicated by question seven. Conscious of the possible implications (eg, the inability to present proof of payment, the occurrence of official vs unofficial payments, medical interventions being more costly elsewhere and so on), two pragmatic elements have been built in that are important from a project management perspective; (1) the requirement to present proof of payment for reimbursements; and (2) a reasonable budgetary ceiling through the maximum tariff list. Finally, based on this list, combined with an estimate of future NR-SAEs calculated on the amount of events during the trial’s initial phase, a budget projection was made for this policy to take effect. Moreover, it is essential that every participant benefits for the same duration. The timelines and their implications are to be clearly communicated upfront to the participants, to avoid unrealistic expectations after the trial (eg, in case of long-term medical requirements). Ongoing serious medical events at trial termination will be followed up until the event is resolved (possible categories: resolved/recovered, resolved/recovered with sequelae or fatal).
On EC-approval of the algorithm policy on NR-SAEs, part of the trial’s funds were realigned to implement it, both prospectively and retrospectively. This was possible because the initial trial’s budget included a buffer for risk mitigation or contingencies, as is good practice for trials conducted in resource-poor settings. The retroactive and prospective application of the algorithm policy is foreseen when the trial activities restart in November 2021. Leading up to this time point, a structural approach and plan (eg, personnel involved at different stages, process for escalation of ambiguous circumstances that do not fit within the binary format and so on) will be developed, starting with updating the informed consent form (ICF) to include the algorithm policy. When returning to the site, participants will be asked to sign and consent to this new EC-approved ICF. The entire implementation process will be well-documented by the site researchers, followed up closely by the sponsor, and will be evaluated at the end of the trial period. Lessons learnt while using the algorithm will be shared with other stakeholders through a new publication.
Considerations for algorithm policy implementation
The Declaration of Helsinki, issued by the World Medical Association,21 describes in art.15 the right to compensation and treatment of participants who were harmed as a result of research participation. However, issues related to compensation, especially of unrelated medical events, and to AC, remain complex in global health research and clear guidance is lacking. Guideline 6 of the CIOMS stipulates that although AC is not obliged, it should be agreed before the start of the study if and how to provide such care for (prior or newly developed) diseases or conditions.10 The Nuffield Council on Bioethics also considers researchers’ responsibilities for participants who encounter a medical condition unrelated to the disease or condition under investigation whereby a minimum standard of care should be offered.22 Furthermore, it has been observed that the international GCP codes of the WHO and the ICH lack adequate consideration for the challenges faced in LMICs.23–25 Applied to the setting in Boende, with its ‘suboptimal’ healthcare system and general lack of health insurance, the team felt that it had a societal responsibility for the enrolled participants, going beyond the project-related deliverables.
Unfortunately, when access to healthcare is lacking, the prospect of getting AC can become an inducement for people to participate in health research,24 26 27 as clinical trials in LMICs typically provide routine medical care to participants9 who often live on a low income and have limited access to health insurance.4 There is a potential risk of exploitation, if not balanced by adequate protection measures and benefit sharing provisions.26 This demonstrates the complexity of conducting research in resource-poor contexts.
However, it could be argued that it is to the benefit of the study quality that participants stay enrolled and without concomitant conditions, and in this sense AC helps to limit dropout and lost to follow-up. In any case, if a study must be conducted in settings with limited or no affordable healthcare, AC obligations cannot be ignored, nor isolated from other professional or institutional responsibilities, nor assigned to the sponsor and site researchers alone.13 Therefore, our research team reached out to locally active development actors to assess the possibilities to strengthen the local healthcare system in Boende in the long run; while, in the short run, we hope that the publication of this algorithm will foster reflection on AC, and recommend other research groups to plan a transparent approach before a trial starts.
Relatedly, AC should be integrated in clinical research guidelines (particularly for global health research) and in national policies and regulations (particularly in LMICs), where governments and other stakeholders should design contextualised measures and requirements to protect the well-being and the rights of the research participants. Such guidance should be further contextualised by protocol and by research context, taking into account the income of households and the nature (or absence) of health insurance schemes. Therefore, AC provisions should be added to the trial protocol, and reviewed by ECs.28 29 This way, ECs will be in a position to assess the fairness and adequacy of the proposed measures upfront. Ideally, the (health) needs are mapped by ECs, community and local government representatives and sponsors, whereby expectations and responsibilities of the different parties, both during and after the study, are agreed. This would enhance the responsibility, accountability and transparency of researchers in improving the participants’ circumstances during global health research. Until ECs are empowered in these negotiations, and thus express expectations towards improving the participants’ circumstances, such initiatives will be limited to the benevolent undertakings of researchers and their funding possibilities or constraints.
Although the algorithm and algorithm policy is just one possible and study-specific way to deal with AC, it could serve as a model for other clinical trials in L(M)ICs or remote areas with access to care problems, provided that it is adapted to the specific research study and context. For example, depending on a country’s healthcare system, gross national product, the demographics of the participants (age, social-economic status and so on), the project funds and so on, co-payment by a participant’s health insurer to cover part of the NR-SAE treatment costs may be included. If this is added, depending on the trial location, for example, in a middle or high-income country (MHIC) with healthcare provision and health insurance, then the modalities and implications need to be assessed upfront, approved by the local EC and monitored during its implementation in practice. However, any expectation of co-payment for medical care in low-income settings is controversial. Co-payment originates from high-income country settings where healthcare is funded via insurance schemes or public funds,30 while in poor communities without health insurance schemes, households can be in the impossibility to contribute the out-of-pocket amount and incur catastrophic health expenditures—being a barrier to Universal Healthcare.4
Despite multiple discussions, extensive literature review and input from social science and ethics experts, there are still some limitations to the algorithm and algorithm policy guidelines. For one, the causality assessment of an SAE to the IP is at times not straightforward. Even if certain AEs can be expected, due to possible concurrent medications or underlying conditions, relatedness cannot always be determined with accuracy.31 Although in the current study causality is evaluated through a binary assessment (‘related’ or ‘unrelated’), a gradual causality assessment (‘possible’, ‘probable’, ‘definite’ and so on) would, to our judgement, not involve implications for the algorithm’s deployment in other clinical trials. For example, each category can, prior to the start of the study, be subdivided into one of the two binary responses.
Furthermore, two local experts affiliated with the PI’s institution have pointed out that the population in DRC regularly turns to traditional medicine for diagnosis, prevention or treatment of physical, mental or social conditions. While the team is conscious of its widespread application, it cannot be covered in the NR-SAE algorithm policy as complementary medicine practices fall outside of the intervention scope. However, its occurrence with regards to NR-SAEs and/or its consequences will be evaluated and if needed will involve further investigation.
Communication with local (health) authorities and participants was an important aspect throughout the implementation and set up of the trial in Boende. Several meetings and workshops were held to ensure the acceptance of the trial and the Ebola vaccine in the community, to understand and appreciate the dynamics of community engagement and to expand the skills of local Communication Task Force Agents involved in the recruitment of participants. Although there was a strong involvement of different local community members during the exploratory phase, the trial set up and further implementation, they were not involved in the algorithm development process. The PI staff, with their expertise and understanding of the local circumstances, acted as local representatives. However, power imbalances between clinicians/researchers and the trial participants in LMICs should also be taken into account. Therefore, as no other community representatives were consulted, this can be considered a limitation of the algorithm policy.
Finally, the NR-SAE policy coverage is limited in time and budget. The WHO Model List of Essential Medicines formed the basis for the Study Pharmacy List. The latter is employed as a reference for the reimbursement of medication costs. In addition, the best care locally available was set as another benchmark for the algorithm policy. As the study team might be confronted with a considerable disability and/or incapacity caused by a NR-SAE, questions rose whether additional care or tools to bring relief to a condition should be offered (eg, counselling, physiotherapy sessions, prostheses and so on). Local experts raised concerns regarding the impossibility to provide care that is simply not available and pointed out the challenges of standard of care. In some cases, proper SAE management could require transfers to other facilities, for example, in the capital. Such interventions would have implications that go far beyond the treatment cost of injuries. While the study cannot take responsibility for what happens outside of the study, the team aims to improve and expand AC and advocates for improved health systems in the study region. Therefore the NR-SAE intervention needs to be the best care locally available, and be sustainable for the participant.
A last important point is that there is dearth of data on the actual costs to implement this kind of policy in vaccine and therapeutic trials in different contexts. We hope that our study will shed some light on these aspects, in order to understand the financial feasibility for (local) research groups that cannot count on significant external funds; and to support advocacy vis-à-vis research funders about the need to systematically include a budgetary line for AC, which would empower both commercial and non-commercial sponsors to provide optimal healthcare to research participants.
Conclusion
The algorithm presented is a practical answer to non-arbitrarily address an AC need in the field. It aims at setting a general framework for support of NR-SAEs and at guiding the decision-making process for each case. Although developed during the trial activities, it anticipates a prospective response to future NR-SAE needs, alongside a retroactive application for preceding cases. Given that this is a first attempt at providing guidance on AC, the algorithm and algorithm policy need to be reassessed at the end of the Ebola vaccine trial. While specific to this trial, it may serve as a basis and can be adapted to the specificities of other clinical trials in settings with access to care constraints. By publishing this algorithm and algorithm policy, we aim to contribute to the methodological and ethical debate on AC and strongly recommend that ECs become directly involved in the discussions and expectations arising from it.
Data availability statement
All data relevant to the study are included in the article.
Ethics statements
Acknowledgments
The authors acknowledge the critical review of Professor Dr D. Watson-Jones (London School of Hygiene and Tropical Medicine) on the algorithm’s principle, which was instrumental to its presentation in this manuscript. We acknowledge the great motivation and the reliability of the study site team. This manuscript is dedicated to the memory of Marguerite Mbenga Lolu, part of the study’s nursing team in Boende, who in June 2020 passed away in childbirth.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Handling editor Seye Abimbola
Twitter @RRavinetto
Contributors PVD had the idea of developing an algorithm for NR-SAE support. GL, YL and JDB were involved in the conception of the algorithm and the development of methodology. GL and YL did the visualisation of the algorithm. VM, PM, JM, TMZ and HMM contributed to the validation process of the algorithm policy. GL wrote the original draft; YL, JDB, TMZ, JM, HMM, PM, VM, PV, ST, J-PVG, PVD and RR contributed to writing, review or revision of the manuscript. RR contributed to addressing the ethical challenges encountered in development of the algorithm policy. J-PVG, PVD and HMM were supervising the algorithm development and manuscript writing. J-PVG is responsible for the overall content as guarantor. All authors had final responsibility for the decision to submit for publication. The corresponding author attests that all listed authors meet authorship criteria and that no others meeting the criteria have been omitted.
Funding This project received funding from the Innovative Medicines Initiative 2 Joint Undertaking (IMI2 JU) under grant agreement No 800 176. This Joint Undertaking receives support from the European Union’s Horizon 2020 research and innovation programme, European Federation of Pharmaceutical Industries and Associations (EFPIA) and the Coalition for Epidemic Preparedness Innovations (CEPI).
Disclaimer The views expressed in this paper are those of the authors and do not necessarily reflect the official positions or policies of other consortium members or funders.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.