Article Text

Statistics from Altmetric.com
Summary box
What is already known about this subject?
Human coronaviruses are known to cause respiratory re-infections, regardless of pre-existing humoural immunity.
There is evidence suggesting that severe acute respiratory syndrome coronavirus type 2 (SARS-CoV-2) had been circulating in Italy before the first COVID-19 case was detected in the country.
What are the new findings?
Prior infections with SARS-CoV-2 (or other viruses/coronaviruses) may arguably predispose to more severe forms of the disease following re-infection with SARS-CoV-2, with an immunological mechanism known as Antibody-Dependent-Enhancement, already observed with infections sustained by other coronaviruses (MERS-CoV and SARS-CoV) or other viruses such as the West Nile Virus and Dengue.
What are the recommendations for policy and practice?
If confirmed by in vivo studies, this hypothesis may have relevant implications for the treatment of severe forms of COVID-19, yet the possibility to produce an effective vaccine against SARS-CoV-2 might be hampered.
The ongoing COVID-19 pandemic, caused by the novel severe acute respiratory syndrome coronavirus type 2 (SARS-CoV-2), has affected 212 countries worldwide at various degrees as of 8 May 2020.1
In this paper we discuss a hypothesis that prior viral infections—either by SARS-CoV-2 or different strains of coronaviruses, or potentially even other respiratory viruses—may predispose to more severe forms of COVID-19, following a secondary infection with SARS-CoV-2.
Most COVID-19 infections are asymptomatic or manifest with mild to moderate respiratory symptoms (fever, cough, sore throat, myalgia, fatigue and even non-severe pneumonia). Of patients with COVID-19, 14%–15% develop severe pneumonia and 5%–6% a critical condition requiring admission to intensive care unit (ICU).2–4 Death may eventually occur after an average of 17.8 days since the onset of symptoms.5
Among all countries, Italy (which was the first European COVID-19 cluster) presents a critical disease pattern as of 8 May 2020, having the third highest number of COVID-19 cases in the world after the USA and Spain, the fourth highest prevalence of the disease after Spain, Belgium and the USA, the third highest total number of deaths attributed to COVID-19 after the USA and the UK, and the third highest prevalence of COVID-19 mortality after Belgium and Spain, despite a current 1% rate of severe/critical disease among active cases, which has been progressively decreasing over time.1
The cross-country discrepancies in the burden of COVID-19 observed so far across the globe cannot be explained only by differences in population age structures.6–8 In fact, Japan has a population double that of Italy, with the proportion of subjects older than 65 being 28.8% in Japan vs 21.7% in Italy.9 10 Nonetheless, as of 8 May 2020, the difference in COVID-19 prevalence between Japan (122 per million) and Italy (3570 per million) is massive.1 Likewise, in Germany the percentage of individuals >65 is reportedly 22.1% (hence slightly higher than Italy), but the prevalence of COVID-19 is currently 2022 per million.1 11 In Iran the proportion of people >65 is 5.5% (hence much younger than the Italian, German and Japanese populations), but the prevalence of COVID-19 is 1246 per million, as of 8 May 2020.1 12
The mortality rate for COVID-19 is reportedly enhanced by 5.6%–10.5% in the presence of any comorbidities (hypertension, diabetes, cardiovascular diseases, cancer and/or chronic respiratory conditions) and becomes significantly and progressively higher after 50 years of age,4 6 although the severe form of the disease increases linearly at any age stage.5 Cold dry weather is a recognised risk factor for respiratory infections, rendering viruses as influenza more stable and individuals more susceptible.13 14 This applies also to SARS-CoV-2, the viability and transmissibility of which reportedly reduce with hot and humid weather conditions.14 Moreover, unfavourable disease progression and clinical outcomes of COVID-19 were found to be associated with cigarette smoking in a systematic review.15
A number of factors may have contributed to enhancing the risk of infection with SARS-CoV-2 in Northern Italy. The age by which half of all young people leave parental home is higher in Italy than other European Union countries,16 and such multigenerational cohabitation probably contributed to increase COVID-19 contagion among elderly individuals. The universal use of face masks was initially discouraged in Italy in order to preserve the limited supplies of personal protective equipment for professional use in healthcare settings; another argument initially was that face masks are ineffective in protecting against coronavirus infections.17 Further major findings of the relevant literature have been summarised in figure 1. An extraordinary elevated incidence of COVID-19 could have been the result of a perfect storm triggered by multiple interplaying factors. The affected areas in Northern Italy (regions of Lombardy, Emilia-Romagna, Piedmont and Veneto) are characterised by high population density18 and recognised air pollution,19 20 especially from fine particulate matter (PM2.5), which was found to increase the risk of death from COVID-19 in the USA.21 Northern Italy includes several cities which, similarly to Philadelphia (USA) during the Spanish flu pandemic in 1918,22 are historically important and densely populated, where social gatherings as well as business activities are certainly fundamental—the latter being vital to the economy of the entire country. These cultural and social dynamics might have influenced the initial resistance and reluctance of the general population to comply with the social restrictions progressively enforced by the Italian government (until full lockdown was established on 21 March). Moreover, the intense case finding in Italy was preceded by an initial overall underestimation of the COVID-19 threat by the Italian government and subsequently by the general population, who perceived the disease as just some sort of influenza, despite worrying news from the first affected country (China).23 Thereafter SARS-CoV-2 was also going to spread to other European countries, which have also now been heavily affected by the disease.1

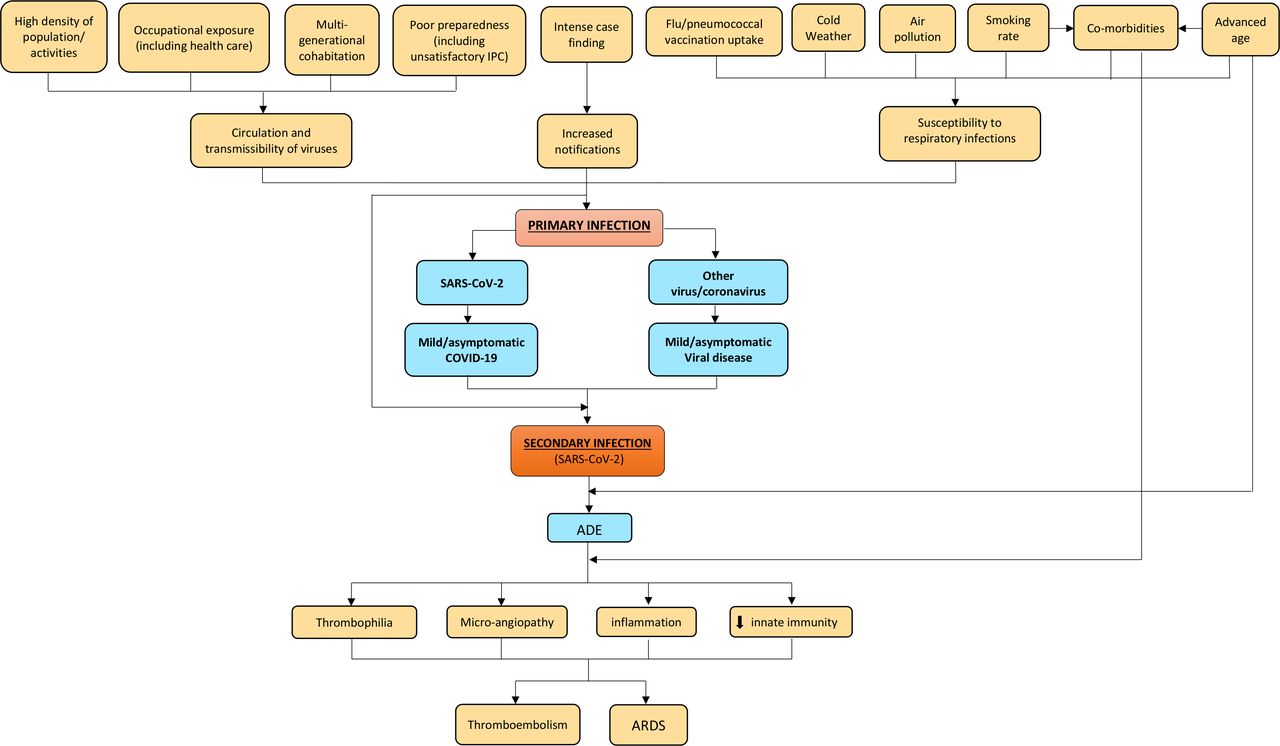{kind=link}
Conceptual framework explaining the relationships between various factors and incidence and severe/critical form of COVID-19. Established items: orange boxes; hypothetical items: blue boxes. ADE, antibody-dependent enhancement; ARDS, acute respiratory distress syndrome; COVID-19, coronavirus infectious disease 2019; IPC, infection prevention and control; SARS-CoV-2, severe acute respiratory syndrome coronavirus type.
The epidemiological investigations conducted by the Italian National Institute of Health (Istituto Superiore di Sanità) suggest that the vast majority of cases but the first three acquired the infection in Italy. It can therefore be reasonably argued that SARS-CoV-2 had been circulating in the country—especially in Lombardy, Emilia-Romagna, Piedmont and Veneto regions—for weeks before the first patient was found.24 Human coronaviruses are known to cause respiratory reinfections regardless of pre-existing humoural immunity, both at the individual and community level.25 At the same time, presumed hospital-associated transmission of SARS-CoV-2 has been reported since the initial stages of the COVID-19 outbreak in Wuhan (China) in 41% of the total number of patients, 70% of whom were healthcare staff.26 This could have also occurred in Italy, where healthcare workers reportedly make up 9% out of all COVID-19 cases.27 We therefore hypothesise (see blue boxes in figure 1) that repeated cycles of infection within a community (especially in older adults)—or even more worryingly in a healthcare setting—could have the potential to cause more severe forms of COVID-19, with acute respiratory distress syndrome (ARDS), the fundamental pathophysiology of severe viral pneumonia due to COVID-19, requiring admission to ICU.28 ARDS associated with COVID-19 shares clinical features with the critical form of the 2003 severe acute respiratory syndrome coronavirus (SARS-CoV) epidemic, in particular lymphopaenia, massive infiltration of phagocytes and inflammation sustained by cytokines.4 8 29
A plausible mechanistic hypothesis could be the antibody-dependent enhancement (ADE), sustained by prior exposure to SARS-CoV-2 or other viruses/coronaviruses.8 Previous circulation and exposure to other coronaviruses similar to SARS-CoV-2 causing mild/asymptomatic cold-like symptoms shall in fact not be ruled out.8 Binding and neutralising antibody response against other types of human coronaviruses was recently reported to increase with age in adult patients,25 and this may explain the increased linear risk of severe COVID-19 with age, with death being significantly higher in patients older than 50.5
Non-neutralising, subneutralising or even fully neutralising antibodies may play a key role in ADE.30 Wan et al30 have recently described a molecular mechanism for ADE involving the Middle East respiratory syndrome coronavirus (MERS-CoV), similar to what is already known for SARS-CoV and flaviviruses as Dengue and the West Nile virus.31–35 While the entry of SARS-CoV into the phagocytes occurred principally through the human ACE2 receptor, the ADE mechanism was shown to be enhanced by antibodies specific for the spike (S) envelope glycoprotein binding with the macrophage receptor and subsequent enhancement of target cell infections.33–35 Likewise, the antibody/opsonised SARS-CoV-2 particles may bind avidly with the IgFc receptors of target cells, increasing the virus yield as well as the production of cytokines. This may also explain the higher risk of thromboembolism reportedly associated with severe/critical COVID-19.4 36 37
Anecdotal clinical reports of ‘biphasic infection’ and ‘cytokine storm’ seem to possibly point towards this direction, biphasic infection simply being the immunological result of a secondary infection by other coronaviruses or a reinfection due to SARS-CoV-2.38–41 An early elevation of serum proinflammatory cytokines, suggesting a pathological mechanism mediated by cytokine storm, has been observed with both severe forms of SARS-CoV and MERS-CoV infections. The latter two viruses share a genomic similarity of about 79% and 50% with SARS-CoV-2, respectively.41 42 Several Rhinolophus bats-related coronavirus strains have been found to share even higher sequence homology with SARS-CoV-2.33 An abnormal humoural response due to ADE, in the early stages of a secondary infection by SARS-CoV-2, may delay the innate antiviral immune response relying on the production of type 1 interferon (IFN-1). This would compromise the initial antiviral response of the host, with subsequent elevated influx of proinflammatory cytokines, hyperinflammatory neutrophils and monocytes-macrophages and hypercoagulable state accountable for ARDS and typical pneumonia observed in patients affected by severe/critical COVID-19 (figure 1).4 41 43 44
If confirmed, this hypothesis would have relevant implications for the treatment of COVID-19 and the development of an effective vaccine. The licensing of a vaccine against human coronaviruses has failed thus far, partly because immunised individuals could potentially be at higher risk of ADE sustained by facilitated uptake of viral antigen-antibody complexes by target cells.4 31 33 44 The approval of a vaccine against SARS-CoV-2 may encounter similar obstacles. Likewise, herd immunity would not be a possibility with COVID-19. WHO recommends passive immunotherapy when vaccine and antivirals are not available for emerging infections.45 In a preliminary uncontrolled case study on five critically ill patients with COVID-19 who developed ARDS, the administration of convalescent plasma—drawn from five patients who recovered from COVID-19 and containing SARS-CoV-2-specific neutralising antibodies (IgG)—between 10 and 22 days since admission significantly improved the clinical status of all, resolving ARDS in four of them within 12 days since transfusion.46 On the other hand, the treatment of severe COVID-19 may also benefit from monoclonal antibodies targeting proinflammatory cytokines4 as well as supplements of IFN-1 in combination with other antiviral drugs.47–50
Whether secondary infections from other coronaviruses or repeated community reinfections of SARS-CoV-2 may account for more severe presentations of COVID-19 observed in some countries compared with others,8 and whether it is only a matter of time for the virus to circulate and infect a significant proportion of the population before causing reinfections and therefore more severe clinical features, are something which will require more indepth epidemiological and immunological/serological investigations. A better understanding of any underlying immunological mechanism or any additional risk factor which could explain cross-country differences in the rates of severe disease and mortality attributable to COVID-19 will help to guide international public health responses during this ongoing pandemic. It will be important to clarify if the ARDS mechanism responsible for the severe respiratory infection could potentially be attributable to ADE.
Two different in vivo strategies could be employed to endorse this hypothesis.
First (observational design), all healthcare workers and blood donors should undergo serum test for COVID-19. Individuals presenting SARS-CoV-2 IgG antibodies should be included in a local/regional/national ad-hoc register and monitored over time for the possible development of severe disease sustained by ADE, which would need to be confirmed by blood count, dosage of IFN and proinflammatory cytokines, in addition to chest CT scan. The risk of developing severe COVID-19 should be estimated and stratified by relevant risk factors, including baseline serum level of SARS-CoV-2-specific IgG antibodies, age, sex, potential occupational exposure, medical history (particularly previous infections and vaccination status), any comorbidities, area of residence and health status of household members, among others.
Second (experimental laboratory design), animal models (hamsters, rodents, palm civets, monkeys, ferrets)33 51 52 could be infected by SARS-CoV-2 (or other viruses/coronaviruses) and subsequently re-exposed to SARS-CoV-2 to verify the possibility of onset of ARDS sustained by ADE.
References
Footnotes
Handling editor Seye Abimbola
Contributors LC, JP, SB, GP and GM equally contributed to conceiving the idea and drafting the manuscript. SC and GS contributed to drafting the manuscript.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests None declared.
Patient consent for publication Not required.
Provenance and peer review Not commissioned; externally peer reviewed.
Data availability statement There are no data in this work.