- Henk DFH Schallig1,
- Halidou Tinto2,
- Patrick Sawa3,
- Harparkash Kaur4,5,
- Stephan Duparc6,
- Deus S Ishengoma7,
- Pascal Magnussen8,9,
- Michael Alifrangis8,9,
- Colin J Sutherland5,10
- 1 Department of Medical Microbiology—Parasitology Unit, Academic Medical Centre, Amsterdam, The Netherlands
- 2 Institute for Health Sciences Research—Clinical Research Unit (IRSS-CRUN), Nanoro, Burkina Faso
- 3 International Centre for Insect Physiology and Ecology, Mbita Point, Kenya
- 4 Clinical Research Department, Faculty of Infectious & Tropical Diseases, London School of Hygiene & Tropical Medicine, London, UK
- 5 Immunology & Infection Department, Faculty of Infectious & Tropical Diseases, London School of Hygiene & Tropical Medicine, London, UK
- 6 Medicines for Malaria Venture, Geneva, Switzerland
- 7 National Institute for Medical Research, Tanga, Tanzania
- 8 Department of International Health, Immunology and Microbiology, Centre for Medical Parasitology, University of Copenhagen, Copenhagen, Denmark
- 9 Department of Infectious Diseases, Rigshospitalet, Copenhagen, Denmark
- 10 Public Health England Malaria Reference Laboratory, London School of Hygiene & Tropical Medicine, London, UK
- Correspondence to Dr Colin J Sutherland; colin.sutherland{at}lshtm.ac.uk
- Received 13 April 2017
- Revised 10 July 2017
- Accepted 10 July 2017
Abstract
Background Management of uncomplicated Plasmodium falciparum malaria relies on artemisinin-based combination therapies (ACTs). These highly effective regimens have contributed to reductions in malaria morbidity and mortality. However, artemisinin resistance in Asia and changing parasite susceptibility to ACT in Africa have now been well documented. Strategies that retain current ACT as efficacious treatments are urgently needed.
Methods We present an open-label, randomised three-arm clinical trial protocol in three African settings representative of varying malaria epidemiology to investigate whether prolonged ACT-based regimens using currently available formulations can eliminate potentially resistant parasites. The protocol investigates whether a sequential course of two licensed ACT in 1080 children aged 6–120 months exhibits superior efficacy against acute P. falciparum malaria and non-inferior safety compared with standard single-course ACT given to 540 children. The primary endpoint is PCR-corrected clinical and parasitological response at day 42 or day 63 of follow-up. Persistence of PCR-detectable parasitaemia at day 3 is analysed as a key covariate. Secondary endpoints include gametocytaemia, occurrence of treatment-related adverse events in the double-ACT versus single-ACT arms, carriage of molecular markers of drug resistance, drug kinetics and patient adherence to treatment.
Discussion This protocol addresses efficacy and safety of sequential ACT regimens in P. falciparum malaria in Africa. The approach is designed to extend the useful life of this class of antimalarials with maximal impact and minimal delay, by deploying licensed medicines that could be swiftly implemented as sequential double ACT by National Malaria Control Programmes, before emerging drug resistance in Africa becomes a major threat to public health.
- malaria
- randomised control trial
Key questions
What is already known on this topic ?
Although still effective, there is increasing concern that antimalarial treatment with artemisinin-based combination therapy (ACT) is seriously threatened by emerging drug resistance.
What is the purpose of this protocol?
This protocol outlines an open-label, randomised three-arm clinical trial in three African settings representative of varying malaria epidemiology to evaluate the efficacy and safety of two sequential ACT regimens as a strategy to prevent persistence of Plasmodium falciparum parasites in treated malaria patients.
Recommendations for policy
The proposed study aims to provide the evidence base for rapidly implementing sequential double ACT treatment if resistance emerges in Africa.
Background
Artemisinin-based combination therapy (ACT) is currently the preferred option to treat uncomplicated falciparum malaria.1 Emergence in the greater Mekong subregion of new genotypes of Plasmodium falciparum, characterised by sequence variants of the P. falciparum kelch 13 propeller domain protein and reduced susceptibility to artemisinin in vivo and in vitro, is a major concern for global public health.1–3 Although it has not yet been documented that such parasites occur in Africa,4 5 recent data from Burkina Faso and Uganda show diminished efficacy of certain ACT combinations.6–8 Furthermore, declining responsiveness of P. falciparum to ACT has been reported from Kenya, linked to submicroscopic persistence of P. falciparum and subsequent parasite recrudescence weeks after apparently successful treatment of clinical malaria with ACT.9 10 These persistent parasites bear clear signals of selection on a number of genes, but not Pfk13.5 11 12 The recent observations of dihydroartemisinin–piperaquine (DHA-PIP) treatment failure in Asia, and in one African isolate, emphasise the urgency of response, as DHA-PIP is more widely deployed in Africa.13–15 Finally, malaria cases imported from Africa to Europe that failed to clear following standard treatment artemether–lumefantrine (AL) have also recently been reported.16 17
Currently, there are no novel treatment regimens available in the global drug development portfolio that could completely replace ACT should a fall in efficacy occur widely. Therefore, it is crucial to preserve the current drugs as long as possible.18 Studies in Cambodia with prolonged artemisinin treatment demonstrated efficacy in areas where standard 3-day treatments are failing, and this may provide an interim solution to preserve ACT efficacy long enough for novel drugs to ‘catch up’.19–21 For falciparum malaria in Africa, prolonged artemisinin treatment may prevent submicroscopic parasite persistence beyond day 3. We propose that sequential administration of two ACTs with different partner drugs is a parsimonious way to achieve prolonged artemisinin treatment.18 22 The rationale behind this proposal, ‘sequential double ACT’, is:
efficacy in preventing persistent parasitaemia should be maximised because following the extended 6-day exposure to artemisinin, parasites will continue also to encounter both partner drugs. This is exemplified in Figure 1, which models component pharmacokinetics when a full course of AL is followed by a full course of DHA-PIP;
transmission potential should be minimised, by the same argument;
safety risks are minimised because artemisinin administered for up to 7 days has an acceptable safety profile, but few partner drugs have been evaluated for safety beyond 3 days; sequential use of two well-tolerated ACTs means no single partner drug is given for more than 3 days;
implementation can be expedited if necessary, as the stringent regulatory requirements prerequisite for new treatments will not apply.
An additional benefit of sequential double ACT is that partner drugs known to exert opposite genetic selection on ACT-resistance-associated loci can be juxtaposed.23
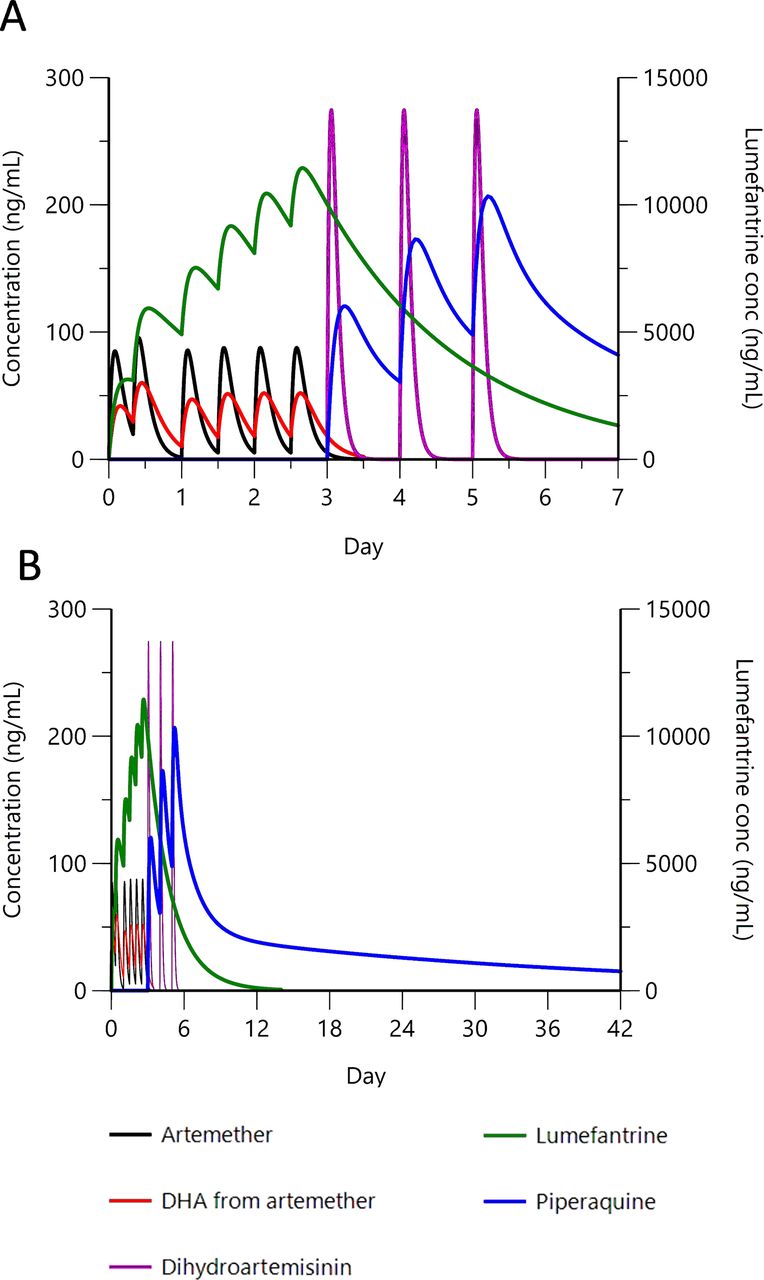
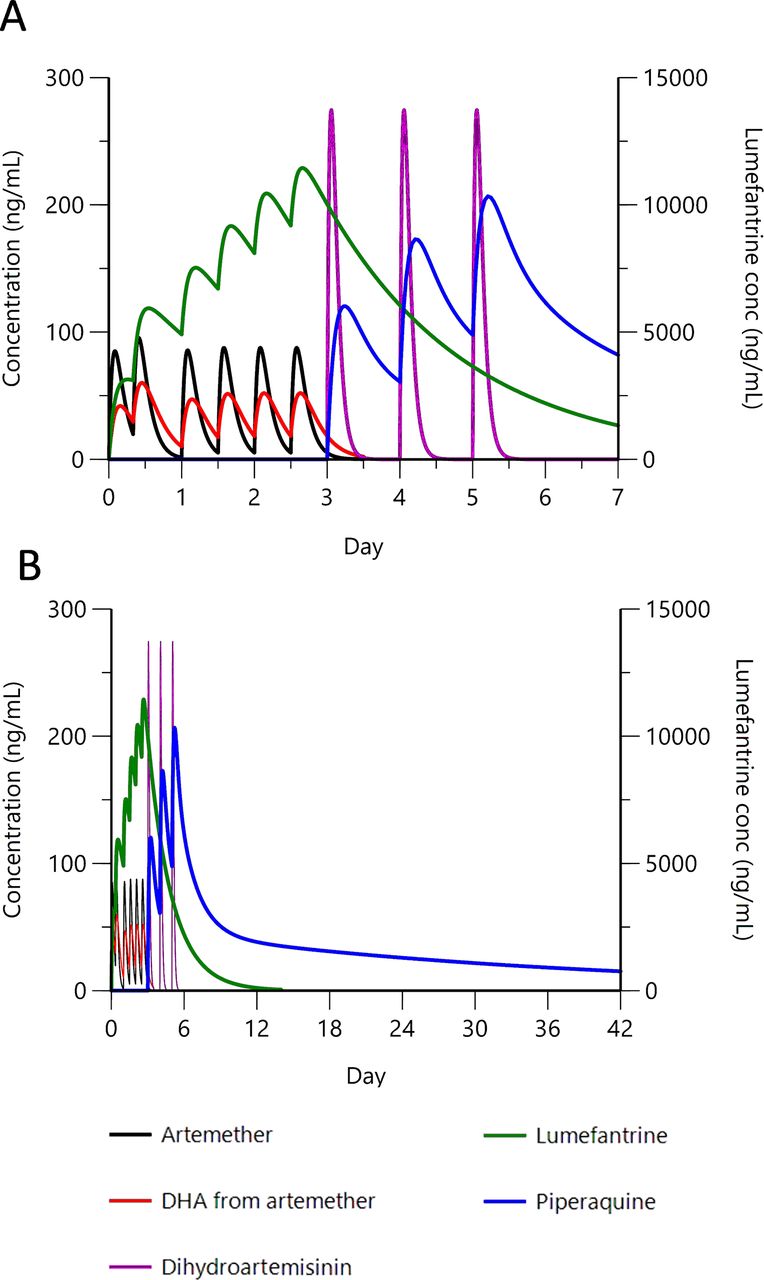{kind=link}
Clinical trial time line: modelling component drug pharmacokinetics for sequential double ACT treatment. Pharmacokinetic representation of the rationale for sequential double ACT. Artemether–lumefantrine followed by DHA is depicted as an example. Pharmacokinetic profiles for (A) 0–7 days and (B) 0–42 days after first dose. Artemether (80 mg x 6, 0–60 hours) and resulting DHA, lumefantrine (480 mg x 6, 0–60 hours), DHA (80 mg x 3, 72–120 hours) and piperaquine (960 mg x 3, 72–120 hours). Peripheral concentration for lumefantrine is shown on the right y-axis; all other compounds on the left y-axis. Artemether is rapidly metabolised to DHA, but both components have antiparasitic activity.38 Modelling parameters are based on published figures for combination-treated adults.39 40 ACT, artemisinin-based combination therapy; conc, concentration; DHA, dihydroartemisinin.
Design and methods
Trial design
To investigate the efficacy and safety of sequential double ACT over 6 days for treatment of uncomplicated P. falciparum malaria in African children, open-label randomised clinical trials will be performed in three malaria epidemiologically representative African settings:
perennial, moderate P. falciparum transmission (Lake Victoria hinterland, Kenya)
high seasonal P. falciparum transmission (West African Sahel, Burkina Faso)
low-intermittent perennial transmission (East African lowland savannah, Tanzania).
Children (6 months to 10 years) meeting defined enrolment criteria will be randomised into three arms:
Arm 1: First-line ACT, according to government guidelines in the participating country. In Kenya and Tanzania: AL; in Burkina Faso: artesunate–amodiaquine (ASAQ);
Arm 2: First-line ACT, according to government guidelines in the participating country, followed by the approved second-line ACT line. In Kenya and Tanzania: AL followed by DHA-PIP; in Burkina Faso: ASAQ followed by AL;
Arm 3: The same regimens as in arm 2, but in reverse order (second-line ACT followed by first-line ACT).
Dosage in all arms will follow the WHO guidelines for each regimen.24
Enrolment criteria follow previous studies,25 26 but can be summarised as follows:
Inclusion: children 6–120 months old attending the health facilities with microscopy-confirmed uncomplicated falciparum malaria; asexual parasite densities between 2000 and 200 000/µL; body weight >5 kg; fever (axillary temperature ≥37.5°C) or history of fever in the preceding 24 hours.
Exclusion: signs of severe malaria; haemoglobin (Hb) <7 g/dL; acute malnutrition (weight for height <70% of the median National Center for Health Statistics/WHO reference); any other concomitant illness or underlying disease; contraindication for trial drugs.
Objectives
The trial has the following study objectives.
Primary study objective
Investigate, using a superiority analysis, whether sequential use of the first-line ACT followed immediately by an established second-line ACT, in either order of use, is more effective in reducing patent or submicroscopic parasite recurrence than is the first-line regimen alone; day 3 quantitative (qPCR) positivity will be analysed as a key covariate in each arm.27
Secondary objective
Investigate, using a non-inferiority analysis, whether sequential use of two ACT regimens leads to a difference in frequency of drug-related adverse events (AEs) compared with the first-line regimen alone.
Exploratory objectives
assess efficacy of the proposed ACT regimens on (sub)microscopic transmissible gametocytes;
identify persistent parasitaemia at days 28 and 42 (and day 63 for low transmission setting) by parasite DNA detection methods;
evaluate genotypes of persisting parasites at loci of interest: Pfk13, Pfcrt, Pfmdr1, Pfap2mu, Pfubp1 and any subsequently validated markers for drug susceptibility;
compare kinetics of artemisinin and partner drugs among patients in the three study arms;
assess acceptability and adherence to each regimen by health practitioners, patients and caregivers.
Sample size
The sample size for this trial is based on a previous study showing that 30%–40% of Kenyan patients had submicroscopic parasite persistence at day 3 determined by qPCR.10 27 Therefore, for every 150 children receiving a single ACT, we expect that 45–60 children will have persistent day 3 parasitaemia by parasite DNA detection, of whom about 20 will also have recurrent microscopically determined parasitaemia at day 28 or 42 (or day 63 for low transmission setting). Thus, 13.3% of the original enrolment will be both day 3 positive by qPCR and day 28/42 positive by microscopy. Using a conservative prediction that 60% of recurrent parasitaemia at day 28/42 will be in those with day 3 subpatent carriage (compared with 84% previously observed in Kenya10), we hypothesise that in the pooled arms receiving two sequential ACTs, this proportion will be reduced to 20%, compared with 60% in the single-ACT arm. This efficacy-based sample size yields excellent power for the primary endpoint even after stratification by day 3 PCR positivity (α=0.05—two-sided; power: 0.9998); we plan to enrol 180 children per arm (to compensate for 20% lost to follow-up). At 80% power, we can detect a smaller reduction from 60% recurrence at day 28/42% to 34%. With the secondary endpoint of PCR detection of recurrent parasitaemia at day 28/42, the power of these analyses is increased further. Total sample size for the three sites will therefore be 540 (single ACT) vs 1080 (sequential double ACT in different combinations), giving substantial power for pooled (secondary) analyses. To summarise:
for an effect size of 40% reduction in recurrent parasitaemia, at α=0.05, >99.9% power is achieved with group sizes of 540 (single ACT) vs 1080 (sequential double ACT, combined)
for 80% power, at α=0.05, an effect size of 26% reduction in recurrent parasitaemia would be detected with these group sizes
In each site, 360 recipients of sequential double ACT will be monitored for AEs, compared with 180 participants on standard ACT. Cardiotoxicity is of general safety concern for ACT regimens, and in a multicentre African study, it was found that prolonged QTc interval occurred in 2.5%–2.6% of ACT-treated children.28 Using this baseline estimate for our standard treatment group, in the pooled analysis of all 1720 participants, we have 87.7% power to detect an increase in prevalence of prolonged QTc to 6% in the combined sequential double ACT groups; with 20% loss to follow-up, power of 79.9% to detect this increase is achieved. Exploratory analyses, such as impact on QTc interval of the order of drug administration, cannot at this stage be evaluated for power as reliable estimates of effect size are lacking.
Clinical investigations and laboratory analyses
Clinical investigations will follow the principles of good clinical practice. During the trial, the following assessments and sample collections will be performed:
Day 0: Primary diagnosis and enrolment of eligible patients providing written informed consent, pretreatment blood sample for microscopy, DNA/RNA analysis and drug level measurements (to test for prior antimalarial use)
Day 0–5: Observed administration of all study drugs; questionnaire for AEs
Day 1–6: Clinical assessment; finger-prick blood sample for blood film, filter papers for molecular analyses and drug measurement
Day 7, 14, 28, 42: Finger-prick for blood films, filter papers for molecular analyses and drug measurement; questionnaire for AEs (In low transmission settings: also day 63)
Passive follow-up: Clinical assessment; finger-prick for blood film, molecular analyses and drug measurement at any unscheduled presentation; questionnaire for AEs.
Clinical specimens will be stored under standard conditions prior to shipment to the appropriate research laboratories. Analyses will follow the principles of good clinical and laboratory practice.29–31 These will comprise:
Efficacy of the intervention
double-read blood film microscopy results
qPCR clearance profiles on days 0, 1, 2, 3, 4, 5 and 7 for every study participant27
genotyping for recrudescence/re-infections using additional timepoints as described10
QT-nucleic acid sequence-based amplification (QT-NASBA) to determine gametocytaemia (days 0–3, 7, 14, 28, 42, and day 63 for low transmission setting)26 32 33
sequence analysis of putative drug resistance loci of interest at day 0 and any subsequent follow-up timepoints with microscopic or PCR-detected persistent parasitaemia.34
Pharmacology
day 0 assessment of blood levels of commonly used antimalarials, relevant to local drug use
pharmacokinetics determined from post-treatment blood samples. Concentrations of all relevant partner drugs (lumefantrine, piperaquine, amodiaquine) will be measured for each site at all timepoints.
Safety assessments
monitoring and recording all AEs and severe AEs (SAEs), by active (at all visits) and passive detection. Any participant with an SAE will be withdrawn from the study and offered remedial treatment or referred to an appropriate facility
monitoring of haematology and blood chemistry, including:
severe anaemia, defined as an Hb <5 g/dL
neutropenia, monitored at day 0, 3, 5 and 7; and also at day 14 and/or 28 in case of neutropenia <1000/mm3
hepatotoxicity, monitored by standard liver function tests (LFTs) at day 3, 7. Cases with LFT >5x upper limit of normal (ULN), or ≥3x ULN for more than 4 weeks, or with other signs, will have additional LFT on day 0 and day 28 samples.
Hy’s law cases: among patients with transaminase levels >3x ULN on day 3 and/or 7, the occurrence of elevated bilirubin (>2x ULN and >35% conjugated) without initial findings of cholestasis (Alkaline phosphatase (ALP) >2x ULN) or other signs of pre-existing liver disease will be monitored and recorded.
routine measurement of vital signs at each visit
monitoring QTc by triple ECG at day 0 pre-dose, day 2 4–6 hours postdose, day 3 4–6 hours after final dose, and on day 28, or when requested by a physician. Data will be corrected using both Bazett’s and Fridericia’s formulae.
Endpoints and outcomes
The primary and main secondary study endpoints derive from the objectives, set out above, thus:
parasitological cure on days 28 and 42 (and day 63 for low transmission setting), stratified by parasite carriage status at day 3
absence of moderate or severe AE during follow-up
haematological and biochemical markers of AE remaining within acceptable range at days 3 and 7.
Additional secondary endpoints include:
gametocyte carriage identified by QT-NASBA at each point during follow-up
qPCR identification of persisting P. falciparum parasitaemia at day 3 and day 7
identification of persisting P. falciparum parasitaemia at days 28 and 42 (and day 63 in low transmission settings only) by microscopy and PCR
selective occurrence of markers of drug resistance in post-treatment samples
estimated adherence to, and acceptability of, proposed treatment schemes.
Ethical considerations
The International Conference on Harmonisation Good Clinical Practice guidelines will be followed. Scientific and ethical review will be sought from local research ethics committees and at appropriate European partner institutions. Study participants will be enrolled only with prior informed consent. Collected data will remain confidential. Study results will be reported in aggregated form; study participants will remain anonymous. Only the clinical team will access identifiable patients’ records. An independent data safety monitoring board will be appointed. The trial design will be registered in an appropriate trial repository.
Discussion
The proposed novel protocol seeks to evaluate efficacy and safety of sequential double ACT for treating P. falciparum malaria patients in Africa, and to test the potential of prolonged ACT regimens against persistent parasitaemia.16 17 21 We have designed this approach to maximise impact, and minimise delay, by deploying licensed medicines that could be implemented in sequential double ACT relatively swiftly, before emerging resistance (should it arise in Africa) becomes a major threat to public health.15–17 22 Based on prior experience in the chosen study sites, we have targeted children with clinical malaria symptoms across a broad age range so as to capture a representative profile of paediatiric malaria in each place.
It has been demonstrated that submicroscopic persistence of P. falciparum within 3 days of ACT treatment is a significant risk factor for subsequent recrudescent parasitaemia post-treatment.10 Use of sequential double ACT is suggested to reduce or prevent the persistence of parasites surviving treatment.17 18 22 Data from extended duration artesunate monotherapy trials,20 35 and the multicentre TRAC study,19 have shown that longer duration of artemisinin treatment, but not higher dosage, is efficacious against slow-clearing P. falciparum genotypes associated with reduced drug efficacy in the Greater Mekong subregion. A short course triple combination regimen (triple ACT) is currently under trial in that region.36 However, our expectation is that extended ACT treatment is more likely to prevent parasite persistence and transmission after treatment. Development of new antimalarial drugs is crucial but, to the best of our knowledge, no newly licensed regimens are ready for widespread deployment in Africa in the next half-decade. Sequential double ACT could be rapidly deployed as a second-line option in African countries, extending the lifetime of current ACT as the drug development pipeline generates new antimalarials in due course. However, the safety profile of the proposed extended ACT regimens is unknown; our study is designed to provide comprehensive data on the occurrence of AE. From these data, we would derive a composite safety endpoint, encompassing the range of events observed, for future studies.
Should our study find that sequential double ACT is well tolerated, and effectively reduces or prevents recurrent parasitaemia and transmission in African settings, this approach should be considered as a legitimate option for treatment of uncomplicated malaria in scenarios where drug resistance is a threat to first-line malaria treatments. Such scenarios would include:
communities in which the average time between clinical malaria episodes receiving ACT has measurably fallen to less than 4 weeks
hospital settings in which severe malaria admissions are increasing in number, suggesting failure of the malaria drugs supplied by the surrounding primary healthcare providers
districts with documented cases of treatment failure, in which health authorities wish to undertake a concerted effort to eliminate or reduce the prevalence of drug-refractory P. falciparum
We do not envisage that sequential double ACT would become first-line policy, but rather a second-line strategy under these scenarios. However, the potential impact of full-scale implementation of sequential double ACT as a first-line therapeutic strategy could also be explored. Such a strategy will increase the costs of malaria treatment, and may also suffer from poor adherence to and acceptability of a 6-day treatment course, notwithstanding the 7-day quinine treatment that is the current rescue policy in cases of ACT failure. Therefore, we would advocate a modelling analysis (not included in the current proposal) linked to our study to evaluate the cost–benefit ratio of sequential double ACT under different conditions of transmission intensity and frequency of post-treatment parasitaemia. Varying levels of effort to ensure good compliance with the sequential double ACT regimens could be tested in the model. Similarly, the optimal order of administration of sequential double ACT can be explored using in silico approaches such as the Simcyp platform.37 On this basis, the project will initiate development of treatment guidelines for implementation of sequential double ACT under each of the indicated scenarios, together with modelling outputs designed to inform authorities as to the prevailing conditions of transmission intensity and drug efficacy under which cost–benefit considerations favour implementation. This will be integrated into our ‘decision tree’.
An alternative output from our study may be the finding that, unlike the situation in Southeast Asia, post-ACT persisting parasites in African malaria patients are not susceptible to regimens of extended duration. This would be an important finding, as it would further emphasise the great urgency needed in the search for novel antimalarials, the development of effective vaccines and continued improvement in non-chemotherapeutic malaria control approaches.
Acknowledgments
We thank colleagues from various institutions for helpful conversations as we developed these ideas. Lene Alifrangis is kindly acknowledged for her assistance with the pharmacokinetic simulations depicted in the figure.
Footnotes
Contributors All authors conceived and developed the proposed trial as a collaborative group. HDFHS and CJS wrote the draft manuscript. All authors have reviewed and approved the final manuscript.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement No additional data are available.
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
References
- 1.
- 2.
- 3.
- 4.
- 5.
- 6.
- 7.
- 8.
- 9.
- 10.
- 11.
- 12.
- 13.
- 14.
- 15.
- 16.
- 17.
- 18.
- 19.
- 20.
- 21.
- 22.
- 23.
- 24.
- 25.
- 26.
- 27.
- 28.
- 29.
- 30.
- 31.
- 32.
- 33.
- 34.
- 35.
- 36.
- 37.
- 38.
- 39.
- 40.